John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Den voksende dokumentation understreger den tætte sammenhæng mellem type II-diabetes (T2D) og Alzheimers sygdom (AD). Det er vigtigt, at disse to sygdomme deler en række patologiske ligheder, herunder amyloidakkumulering, oxidativ stress, inflammation og celledød. Hidtil har der manglet lægemidler til behandling af AD og T2D, og der er et afgørende behov for opdagelse og udvikling af nye terapiformer til behandling af disse sygdomme. En række undersøgelser på mennesker og gnavere har vist, at tilskud af metaboliske hormoner er yderst værdifuldt for at forbedre den kognitive funktion og den generelle metaboliske sundhed i forbindelse med både T2D og AD. Det pancreatiske hormon amylin har vist sig at være en afgørende komponent i sygdomsætiologien ved både T2D og AD, selv om den nøjagtige rolle, som amylin spiller i disse sygdomme, endnu ikke er velforstået. Her gennemgår vi kritisk den aktuelle litteratur, der anvender humant amylin eller dets syntetiske analog, pramlintid, samt amylinreceptorantagonister til behandling af AD.
Indledning
Alzheimers sygdom (AD) er en progressiv, invaliderende neurodegenerativ sygdom, der er karakteriseret ved ophobning af amyloid-beta (Aβ) plaques og neurofibrillære tangles, der består af hyperfosforyleret tau1. Ophobningen af disse patologiske peptider bidrager til underskud i udøvende funktioner som f.eks. indlæring og hukommelse, humør, affekt osv. og udgør en betydelig byrde for patienten og de pårørende. Forekomsten af AD stiger med alarmerende hastighed i USA, og det anslås, at 5,5 millioner amerikanere lever med AD i 2017, og dette tal forventes at blive tredoblet inden 20502. Desuden overstiger omkostningerne til pleje og behandling af AD-patienter i øjeblikket 200 mia. dollars årligt og forventes kun at stige3. Selv om AD helt klart er et monumentalt problem i og uden for USA, er behandlingsmulighederne stadig meget begrænsede4. Der er gennemført mange lægemiddelforsøg med en bred vifte af målrettede metoder, men der er i øjeblikket kun seks lægemidler godkendt af FDA til behandling af Alzheimers sygdom, og de er kun symptomatiske behandlinger5, 6. Hidtil har størstedelen af de farmakologiske midler, der er udviklet, været specifikt rettet mod Aβ- eller tau-patologien, men ingen har haft succes med at fjerne eller forebygge patologien4. Der er således et grundlæggende behov for at udvikle levedygtige terapier og forebyggende behandlinger for AD.
Aldersrelateret (sporadisk) AD er en kompliceret multifaktoriel sygdom, der har mange genetiske og miljømæssige påvirkninger. Miljø og livsstil er stærkt involveret i udviklingen af sporadisk Alzheimers sygdom; faktorer som f.eks. kost7-9, fedme8-10, metabolisk syndrom7, type II-diabetes (T2D)9, 11 og hjerte-kar-sygdomme12 er alle blevet inddraget i årsagsforløbet for Alzheimers sygdom. Det er af afgørende betydning, at antallet af tilfælde af fedme og diabetes stiger hurtigt parallelt med Alzheimers sygdom12, 13. Selv om forholdet mellem fedme og Alzheimers sygdom er noget uklart, er der beviser for, at fedme midt i livet spiller en rolle i udviklingen af Alzheimers sygdom10. Endnu vigtigere er det, at fedme almindeligvis ledsages af en række andre sygdomme, herunder hjerte-kar-sygdomme, forhøjet blodtryk, dyslipidæmi, T2D, slagtilfælde osv.14 . Forekomsten af T2D er hurtigt stigende, og CDC anslår, at ca. 30,3 millioner mennesker (1 ud af 10 voksne) i USA har diabetes og svimlende 84,1 millioner (1 ud af 3 voksne) har prædiabetes, hvoraf de fleste er uvidende om deres tilstand. På grund af et omfattende fald i fysisk aktivitet, der ledsages af en samtidig stigning i fødeindtagelse og dårlig kost, foreslås det desuden, at antallet af fedme, T2D, metabolisk syndrom og hjerte-kar-sygdomme kun vil stige til anslået 600 millioner T2D-tilfælde på verdensplan inden 203515.
Den dokumentation, der involverer metabolisk funktion og sygdom i processen med kognitiv tilbagegang og aldring, er betydelig16, 17. For eksempel rapporterer ca. 70 % af de personer, der er diagnosticeret med T2D, om kognitiv svækkelse, og et betydeligt antal T2D-patienter udvikler senere demens16, 18-21. Personer, der er diagnosticeret med T2D i mindst fem år, har en betydeligt øget risiko for at udvikle Alzheimers sygdom sammenlignet med personer, der har lidt af T2D i mindre end fem år17. Tilsammen tyder disse data på, at den stigende forekomst af T2D i befolkningen kan bidrage til den stigende forekomst af AD.
T2D er oprindeligt karakteriseret ved højt blodglukose og insulin, hvilket fører til hyperinsulinæmi; vigtigt er det, at amylin, et lille stofskiftehormon, der produceres af β-isletceller i bugspytkirtlen, sampakkes og samsekreteres med insulin og således overproduceres ved T2D.22. Det er vigtigt, at der er en række patologiske træk, som er til stede i både T2D og AD: 1) nedsat hjernemetabolisme og metabolisk hormonresistens 2) amyloidpatologi 3) oxidativ stress (OS) og inflammation. Kronisk hyperinsulinæmi og hyperamylinæmi fører til en række fysiologiske problemer: kronisk hyperinsulinæmi fører til insulinresistens i systemet22 , nedsat insulintransport over blod-hjernebarrieren (BBB)23, 24 og dermed nedsat insulinsignalering i hjernen25. Tab af insulinsignalering i hjernen er forbundet med en række AD-relaterede patologiske træk, herunder øget Aβ-produktion, tau-fosforylering og neuroinflammation.
Dertil kommer, at amylin deler lignende patologiske træk med Aβ ved høje koncentrationer26 og kan være en fælles vej mellem de to sygdomme. F.eks. er amylinfibriller fundet i bugspytkirtlen hos 95 % af T2D-patienter27-29 og forårsager en række fysiologiske forstyrrelser, herunder aberrant Ca2+-indstrømning, øget sekretion af proinflammatoriske cytokiner30,31 og i sidste ende β-isletcelletab32. Desuden krydser amylin let BBB og danner amylinfibriller samt blandede plaques med Aβ i hjernen og kan være ansvarlig for AD-lignende patologi og Aβ-sætning i T2D33-35. Amylin er kendt for at påvirke langtidspotentiering (LTP) i hippocampus og kan have en medfødt indflydelse på den kognitive funktion i hjernen36-39. Det er imidlertid fortsat uklart, om amylin er en toksisk insult i disse sygdomme, eller om dets funktionelle tab gennem aggregering eller sent stadie af β-celletab i T2D bidrager til udviklingen af en AD.
Dichotomien om amylinsignalering
Der er stadig stor debat om involveringen af amylinreceptoren (AMYR) og amylinsignalering i sygdomsforløbet og ætiologien af T2D og AD. Den forskning, der har til formål at afdække dette forhold, vokser hurtigt. Al relevant forskning har konsekvent vist, at modulering af amylinsignalering påvirker AD-relateret patologi. Arten af dette forhold er imidlertid endnu ikke blevet konkret belyst. Flere grupper har fremlagt overbevisende data, der tyder på, at amylinsignalering er gavnlig i forebyggelsen af AD-relateret patologi og kognitive underskud både in vivo og in vitro40-44. Det er vigtigt, at pramlintid, en rekombinant ikke-aggregerende form af amylin, der anvendes sammen med insulinbehandlinger til behandling af diabetes og forbedrer den glykæmiske kontrol, reducerer kropsvægten og reducerer serummarkører for OS45-47, også viser sig at være lovende som et AD-terapeutisk middel. Indtil nu har der imidlertid ikke været nogen kliniske forsøg, der har haft til formål at anvende amylin eller pramlintid som et terapeutisk middel til behandling af demens. Klare beviser fra gnaverundersøgelser tyder på, at kronisk behandling med enten humant amylin eller pramlintid udgør en stærk terapeutisk fordel med hensyn til at reducere AD-relateret patologi; amylin/pramlintid-supplementering reducerer niveauer af opløseligt Aβ, plaquebyrde, tau-fosforylering, neuroinflammation og OS, samtidig med at kognitionen forbedres40-42,44. Ovenstående data tyder på, at et tab af medfødt amylinsignalering i CNS på grund af aggregering giver anledning til en øget risiko for udvikling af AD og er dækket mere detaljeret i Grizzanti et al. 201848.
I modsætning hertil viser undersøgelser også, at humant amylin og Aβ har lignende toksiske virkninger, og at disse toksiske virkninger kan afhjælpes ved hjælp af AMYR-antagonist36-39,49. For eksempel viser data, at in vivo-behandling med AMYR-antagonister giver meget lignende fysiologiske fordele som amylin- eller pramlintidbehandling. Behandling af TgCRND8 AD-mus med AC253, en AMYR-antagonist, eller dens cykliske modstykke cAC253 reducerer neuroinflammation, niveauer af opløseligt Aβ og plakatbyrde, samtidig med at kognitionen forbedres50. Tilsvarende viser in vitro/ex-vivo-undersøgelser, at lav dosis humant amylin eller Aβ forårsager forstyrrelser i LTP, og at disse underskud blokeres af AC253 eller pramlintid38,39, og at højere doser humant amylin/amylinoligomerer er forbundet med ukontrolleret Ca2+-influx, som er stærkt forbundet med celledød26,32. Tilsammen understøtter disse data en toksisk funktion af amylinoligomerer og dermed en potentiel terapeutisk mekanisme for AMYR-blokade. I modsætning hertil har andre vist, at de gavnlige virkninger af amylin kan blokeres ved hjælp af AC25341. Det terapeutiske potentiale ved amylinbehandling eller -hæmning er således fortsat uklart og fremhæver den komplekse og dikotomiske karakter af amyloider i hjernen og periferien.
Sammensætning af puslespillet
Der er en række huller i den nuværende litteratur, som skal udfyldes for at give et mere fuldstændigt billede af amylinhistorien: 1) arten af det medfødte amylinsystem og amylinsignalering i hjernen 2) Aβ- og pramlintid-signaleringsevnen gennem de tre vigtigste AMYR-receptorer og beslægtede receptorer 3) de terapeutiske mekanismer, hvorved amylin/pramlintid- eller AMYR-hæmning formidles. For det første viser interessante nye data, at AMYR ikke kun er involveret i signalering, men også i ligandtransport gennem BBB. AMYR er en heterodimær receptor, der består af en calcitoninreceptor og et receptoraktivitetsmodificerende protein (1-3)51 . I den forbindelse reducerede en 50 % global knockdown af calcitoninreceptoren (en nøglekomponent i AMYR) signifikant den mængde AC253, der blev fundet i hjernen50 , hvilket indikerer, at AMYR, der er placeret i BBB, er involveret i transporten af disse ligander ind i hjernen og kan også være involveret i transport af amylin og pramlintid ind i/ud af hjernen. Eksistensen af disse BBB-transportmekanismer tyder på, at amylin sandsynligvis har en medfødt fysiologisk funktion i hjernen, da dets transport ind i hjernen er nøje kontrolleret. Det er imidlertid stadig uklart, hvordan amylinsignalering eller mangel på samme fører til de patologiske træk ved AD, og om AMYR er det middel, hvorigennem Aβ formidler sine toksiske virkninger.
Dernæst findes der modstridende beviser med hensyn til forholdet mellem Aβ og AMYR. Selv om flere undersøgelser klart viser, at humant amylin og Aβ har lignende virkninger på LTP i CNS, og at brugen af AMYR-hæmmere forbedrer disse skadelige virkninger36-39, tyder andre beviser på, at Aβ (1-42) ikke er i stand til at signalere gennem AMYR til at fremkalde nogen form for cAMP-respons ved en lang række forskellige koncentrationer52. Det er muligt, at Aβ aktiverer forskellige signalkaskader gennem interaktion med AMYR eller blot fungerer som en inaktiv konkurrerende hæmmer, men dette er endnu ikke blevet påvist.
Dertil kommer, at en separat undersøgelse viste, at oligomerisk amylin formidler sine toksiske virkninger direkte gennem AMYR og indirekte gennem TRPV4, en ikke-selektiv kationkanal26. Lave koncentrationer af humant amylin fremkalder et Ca2+ -respons, som formidles gennem dets native receptor. Ved højere koncentrationer danner humant amylin imidlertid oligomerer og aktiverer aberrant signalering, der resulterer i aktivering af TRVP4-kanaler og giver mulighed for ukontrolleret kationtilstrømning, især Ca2+. Farmakologisk blokering af AMYR og TRPV4 viser, at begge receptorer er nødvendige for, at oligomerisk humant amylin kan fremkalde sine toksiske Ca2+ -virkninger26. Det er derfor sandsynligt, at Aβ formidler sine toksiske virkninger på AMYR på en lignende måde, selv om der endnu ikke foreligger nogen data herom. Ukontrolleret Ca2+-indstrømning er forbundet med en række patologiske fænomener, herunder ukontrolleret vesikulær frigivelse, OS- og mitokondriel dysfunktion, apoptose osv. I den forbindelse er det sandsynligt, at cellulær dysfunktion og udviklingen af yderligere AD-lignende patologi, der skyldes toksisk amyloid-signalering, formidles gennem både AMYR og TRPV4. Som sådan er det nødvendigt at skelne de signalkaskader, der modulerer forholdet mellem AMYR og TRVP4. Endvidere er der behov for farmakologiske eksperimenter, der anvender Aβ og pramlintid over en bred vifte af doser for at bestemme Aβ og pramlintids virkninger på Ca2+ strømme, LTP, cAMP-produktion og andre signalkaskader for at bestemme deres signaleringsevner. Disse eksperimenter vil bidrage til at udfylde nogle af tomrummene i den nuværende litteratur med hensyn til AMYR og dens involvering i sygdomstilstande (figur 1).
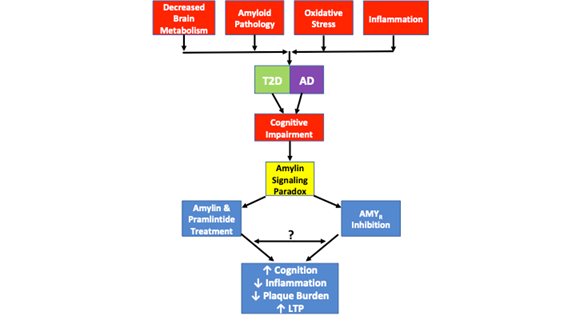
Figur 1. skildrer amylinsignaleringsparadokset og patologiske ligheder, der er observeret i T2D og AD. Nedsat hjernemetabolisme, amyloidpatologi, oxidativ stress og inflammation er alle fælles patologiske træk, der observeres i begge sygdomme. Selv om ikke alle tilfælde af T2D eller AD omfatter hvert af disse patologiske træk, er der i alle tilfælde kognitive forringelser. Amylinsignalparadokset kommer ind i billedet, da undersøgelser har vist, at både AMYR-hæmning og AMYR-agonisme via amylin- og pramlintidbehandling resulterer i forbedret kognition, nedsat inflammation, nedsat plakatbyrde og øget LTP. De signalmekanismer, der styres af AMYR-agonisme og AMYR-antagonisme, er endnu ikke fuldt ud opklaret. Mens amylinsignalering traditionelt er forbundet med cAMP- og PKA-signalering, er det uklart, om andre kaskader også aktiveres af amylin/pramlintid. Desuden er det uklart, om AMYR-antagonister, amylinoligomerer eller Aβ signalerer gennem AMYR, eller om der er nogen ligheder eller krydssammenfald mellem alle disse AMYR-ligander. Som sådan vil en række eksperimenter, der foreslås i denne gennemgang, bidrage til yderligere at belyse AMYR’s sande natur.
Konklusioner
Den nuværende uenighed med hensyn til amylinsignaleringens rolle i hjernen viser et væsentligt behov for yderligere belysning af amylins involvering i både AD og T2D. I T2D er det sandsynligt, at amylin i de tidlige stadier af sygdommen oversvømmer hjernen, danner oligomerer, inducerer aberrant signalering gennem sin oprindelige receptor og rekrutterer TRPV4 for at inducere patologisk Ca2+-tilstrømning, der resulterer i udbredt neuronal dysfunktion, der manifesterer sig som OS, ukontrolleret vesikulær frigivelse og interneuronal dysfunktion, inflammation og resulterende celledød. Denne mekanisme kan være ansvarlig for den indledende overgang fra den sunde hjerne til hjernens aldring ved metabolisk sygdom. Som sådan kan AMYR- eller TRVP4-hæmning på visse tidspunkter i metabolisk sygdom og i de tidlige stadier af diabetes være berettiget for at blokere de toksiske virkninger af oligomerisk amylin eller Aβ. Der er imidlertid også stærke beviser for, at amylinerstatning med enten humant amylin eller pramlintid reducerer de fleste af de vigtigste AD-relaterede patologier og samtidig forbedrer kognitionen i gnaermodeller af AD. Som sådan kan det være berettiget at erstatte amylinsignalering med amylin eller pramlintid i de midterste til sene stadier af diabetes, når amylinsignalering er tabt på grund af aggregering, oligomerisering eller β-celletab. Med henblik herpå er der også behov for at skelne den tidsmæssige præsentation af patologiske hændelser i metabolisk forbundet hjernealdring og terapeutiske muligheder for tidlige, mellemliggende og sene sygdomsstadier. Kritisk analyse og afprøvning af disse amyloiders direkte karakter og signaleringsevne samt den terapeutiske karakter af specifikke tidsmæssige behandlinger kan bidrage til at bygge bro over kløften mellem AMYR-hæmningsterapier og amylinerstatningsterapier.
Funding
Funding til denne artikel blev ydet af National Institutes of Aging grant 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau og synaptisk dysfunktion. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurologi. 2013; 80: 1778-1783.
- Sammenslutning As. 2016 Alzheimer’s disease facts and figures (Fakta og tal om Alzheimers sygdom). Alzheimers & Demens. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil og memantin til moderat til svær Alzheimers sygdom. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glukoseniveauer og risiko for demens. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of dementia prevalence in the United States and China. Obesity. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus og risiko for demens: en metaanalyse af prospektive observationsundersøgelser. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Type 2-diabetes som en risikofaktor for Alzheimers sygdom: de forvirrende faktorer, interaktioner og neuropathologi, der er forbundet med dette forhold. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurologi. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Fedme og de comorbide tilstande i forbindelse hermed. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF diabetes atlas. Bruxelles: International Diabetes Federation. 2013.
- K Dash S. Kognitiv svækkelse og diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risiko for demens blandt personer med diabetes mellitus: en befolkningsbaseret kohortestudie. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Demens og kognitiv tilbagegang i type 2-diabetes og prædiabetiske stadier: mod målrettede interventioner. The lancet Diabetes & endokrinologi. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus og Alzheimers sygdom: fælles patologi og behandling. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prævalens af diabetes og prædiabetes og deres risikofaktorer blandt voksne bangladeshiske voksne: en landsdækkende undersøgelse. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus og risikoen for demens The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insulinnedbrydende enzym regulerer niveauerne af insulin, amyloid β-protein og det intracellulære domæne af β-amyloidprækursorprotein in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulinbinding til hjernens kapillærer er reduceret hos genetisk overvægtige, hyperinsulinæmiske Zucker-rotter. Peptides. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolisme. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Insulinniveauer er nedsat i cerebrospinalvæsken hos kvinder med prodomal Alzheimers sygdom. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neuropharmakologi. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mekanismer, der forbinder fedme med insulinresistens og type 2-diabetes. Natur. 2006; 444; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Forringet glukosetolerance er forbundet med øget islet amyloid polypeptid (IAPP) immunoreaktivitet i pankreatiske betaceller. Det amerikanske tidsskrift for patologi. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Aktivering af NLRP3-inflammasomet ved islet-amyloid-polypeptid giver en mekanisme for øget IL-1β i type 2-diabetes. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatoriske markører og risiko for type 2-diabetes: en systematisk gennemgang og meta-analyse. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimers β-amyloid, human islet amylin og prionproteinfragment fremkalder intracellulære frie calciumforhøjelser ved en fælles mekanisme i en hypothalamisk GnRH-neuronal cellelinje. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal amylinaflejring, peroxidativ membranskade og øget IL-1β-syntese i hjerner fra Alzheimer-patienter med type 2-diabetes og i diabetiske HIP-rotter. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylinaflejring i hjernen: en anden amyloid i Alzheimers sygdom. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo udsåning og krydsudsåning af lokaliseret amyloidose: et molekylært link mellem type 2-diabetes og Alzheimers sygdom. Det amerikanske tidsskrift for patologi. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylinreceptor: et fælles patofysiologisk mål i Alzheimers sygdom og diabetes mellitus. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ)-peptid aktiverer direkte amylin-3-receptorsubtypen ved at udløse flere intracellulære signalveje. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide Antagonizes Beta Amyloid (Aβ)-and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloid-induceret depression af hippocampal langtidspotentiering er medieret gennem amylinreceptoren. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Intraperitoneal injektion af det pancreatiske peptid amylin reducerer potent adfærdsforringelse og amyloidpatologi i hjernen i murine modeller af Alzheimers sygdom. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylinreceptorligander reducerer den patologiske kaskade af Alzheimers sygdom. Neuropharmakologi. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotektive virkninger af amylinanalogen pramlintid på Alzheimers sygdomspatogenese og kognition. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilisering af β-catenin ved mutationer i presenilin-1 potenserer neuronal apoptose. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin-behandling reducerer neuroinflammation og forbedrer abnorme mønstre af genekspression i hjernekorten i en Alzheimers sygdomsmusmodel. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetat injektion til behandling af type 1 og type 2 diabetes mellitus. Clin Ther. 2007; 29: 535-562.
- Singh?Franco D, Perez A, Harrington C. Effekten af pramlintideacetat på glykæmisk kontrol og vægt hos patienter med type 2-diabetes mellitus og hos overvægtige patienter uden diabetes: en systematisk gennemgang og metaanalyse. Diabetes, fedme og metabolisme. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintid som supplement til insulinbehandling forbedrer langsigtet glykæmisk og vægtkontrol hos patienter med type 2-diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Virkninger af β-amyloidprotein på humane neuroner kommer til udtryk gennem amylinreceptoren. Det amerikanske tidsskrift for patologi. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyklisk AC253, en ny amylinreceptorantagonist, forbedrer kognitive underskud i en musemodel af Alzheimers sygdom. Alzheimers & Demens: Translational Research & Clinical Interventions. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reduceret nociceptiv adfærd i islet amyloid polypeptid (amylin) knockout-mus. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Aktivitet af pramlintid, rotte- og humant amylin, men ikke Aβ1-42 ved humane amylinreceptorer. Endokrinologi. 2014; 155: 21-26.