Incidensen og prævalensen af nyrestenssygdom fortsætter med at stige i den almindelige befolkning. Med en livstidsprævalens på op til 10 % er nefrolithiasis (NL) og nefrocalcinose (NC) derfor en stor sundhedsbelastning, især i den vestlige verden (1). NL og NC er forbundet med betydelig morbiditet og udvikling til kronisk nyresygdom på grund af gentagelse, gentagne kirurgiske/endoskopiske indgreb og samtidig inflammation. På et forenklet niveau skyldes nyrestensdannelse en ubalance mellem urininhibitorer (f.eks. citrat, magnesium, uromodulin og pyrofosfat) og promotorer (f.eks. oxalat, calcium, fosfat, urat og cystin) af krystallisering, der overskrider overmætningen med efterfølgende aggregering, nukleation og stenvækst ved Randalls plaque (figur 1). Denne ubalance kan skyldes ændret enteral og/eller renal håndtering af enten promotorer eller inhibitorer, såsom enteral malsekretion af oxalat eller renal malreabsorption af calcium (figur 1) (figur 1).
Figur 1. Ubalance af urininhibitorer og promotorer af krystallisering, der fører til nyrestensdannelse. Koncentrationen af urininhibitorer og promotorer påvirkes og kontrolleres af både tarm- og nyretransportører (GI-nyre-akse). Disse transportører og udvekslere, som f.eks. SLC26A1, er ansvarlige for sekretion og absorption. Med hensyn til oxalat fører enteral hyperabsorption men malsekretion og/eller renal hypersekretion men malabsorption til urinoxalatniveauer, der overstiger overmætningen og derved fremmer krystallisering og efterfølgende stendannelse .
Den underliggende ætiologi for NL menes at være multifaktoriel med en miljømæssig, bemærkelsesværdig kost-, hormonel og genetisk komponent. I tvillingestudier er arveligheden af nyresten blevet anslået til 56 % (3), og op til to tredjedele af hyperkalciuriske stendannere har slægtninge med NL (4). Selv om calciumholdige nyresten udgør mere end 80 % af alle, er det genetiske grundlag for sådanne sten stadig stort set ukendt (5). Bortset fra varianter i CLDN14, TRPV5, SLC34A1, ALPL, CASR og UMOD har genom-dækkende associationsundersøgelser endnu ikke givet væsentlige genetiske faktorer (6-8). Der er imidlertid blevet identificeret risikoalleler inden for gener, som også viste sig at overføre sygdommen på mendelsk basis, såsom CASR, SLC34A1 og SLC2A9 (9, 10). Til dato er mere end 30 enkeltgener med en Online Mendelian Inheritance in Man-defineret fænotype blevet identificeret som værende involveret i NL/NC, hvis de er muteret (tabel 1).

Tabel 1. Gener, der vides at forårsage monogene former af NL/NC.
Arvemåder for monogene former omfatter autosomal-dominant, autosomal-recessiv og X-bunden transmission. Det er interessant, at der for flere af disse gener er rapporteret både recessive og dominerende arvegange: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 og SLC4A1. Mens de fleste syndromiske og alvorlige medfødte lidelser udviser et recessivt arvelighedsmønster (Bartter, Lowe, Dent, FHHNC og distal renal tubulær acidose med sensorineural døvhed), er mildere tilstande snarere forbundet med mutationer i dominerende gener. Størstedelen af de kodede proteiner udgør renale transportører af opløste stoffer (f.eks. SLC34A1, SLC34A3 og SLC9A3R1), men også kloridkanaler (CLCN5), tight-junction-proteiner (f.eks. CLDN16/CLDN19) og metaboliserende enzymer (f.eks. AGXT, APRT og CYP24A1) er blevet fundet defekte hos patienter med NL/NC. Den underliggende defekt er derfor for det meste placeret i selve nyrens tubulære system og kan derfor tilskrives som tubulopati. Omvendt er a priori extrarenale tilstande, som i primær hyperoxaluri (PH), hvor dysfunktion af leverenzymer (AGXT, GRHPR og HOGA1) forårsager oxalatophobning med sekundær nyreaffektion, tænkelige årsager til NL/NC. Selv om hver enkelt sygdomsfænotype menes at repræsentere en forholdsvis sjælden enhed, kan enkeltgen-årsager muligvis tegne sig for et betydeligt antal patienter på grund af deres brede genetiske heterogenitet (42). Ud over den genetiske heterogenitet er der også en allelisk variation, hvor trunkerende varianter snarere resulterer i et funktionstab, og missense-varianter (hypomorfe) kan forårsage ret subtile defekter, som kan overses klinisk, især hos voksne stendannere. Et andet nyligt værdsat fænomen drejer sig om gen-dosiseffekter i flere af de førnævnte nyrestensgener. I SLC34A3 f.eks., der koder for en af de vigtigste fosfattransportører i den proximale tubulus (NaPiIIc), blev det vist, at heterozygote individer ikke længere blot kan betragtes som sunde bærere, da de viser nyrekalkninger og/eller knoglemanifestationer betydeligt hyppigere end wild-type individer; men stadig i mindre grad end bialleliske (homozygote og sammensatte heterozygote) individer (43). Lignende observationer blev rapporteret for familier med mutationer i CYP24A1 (44). Bidraget fra monogene sygdomme til den samlede prævalens af nyrestenssygdomme er ikke blevet undersøgt omfattende tidligere. Især mangler der genetiske beviser baseret på brede screeninger af et væld af forårsagende gener i store patientkohorter. Omfattende genetisk testning har tidligere været for dyrt og ineffektivt. For de fleste personer med NL/NC har det derfor ikke været muligt at foretage mutationsanalyser for en forårsagende genetisk defekt på trods af, at viden om den molekylære årsag til NL/NC kan have vigtige konsekvenser for prognose, profylakse og/eller behandling. Kun grove skøn er blevet udledt af kliniske observationsundersøgelser: på grundlag af en stor dataindsamling af analyser af stensammensætningen blev det konkluderet, at monogene årsager ikke overstiger 9,6 % hos børn og 1,6 % hos voksne (45). I det sidste årti er denne situation imidlertid begyndt at ændre sig med fremkomsten af high-throughput sekventeringsteknikker.
High-Throughput mutationsanalyse hos patienter med NL/NC
For at undersøge patienter med nyrestenssygdom for tilstedeværelsen af patogene mutationer i kendte sygdomsgener etablerede vi et genpanel baseret på mikrofluidisk multiplex-PCR og konsekutiv NextGen sekventering (Fluidigm™/NGS) (46, 47).
I en “pilotundersøgelse” rekrutterede vi efter hinanden 268 genetisk uopklarede personer fra typiske nyrestensklinikker; 102 pædiatriske og 166 voksne probander. Som et resultat heraf identificerede vi 50 skadelige varianter i 14 ud af 30 analyserede gener, hvilket førte til en molekylær diagnose i 15 % af alle tilfælde. I den pædiatriske undergruppe påviste vi en forårsagende mutation i 21 %, mens der blandt voksne var skadelige varianter til stede i 11 % (Figur 2A) (48). Mutationer i cystinuria-genet SLC7A9 blev fundet hyppigst i den voksne kohorte (Figur 2B). To opfølgende undersøgelser kunne bekræfte disse resultater. For det første blev 17 % af tilfældene i en udelukkende pædiatrisk kohorte af 143 NL/NC-patienter forklaret med mutationer i 14 forskellige gener (49). For det andet blev der i en kohorte på 51 familier med alder for NL/NC-manifestation før 25 år anvendt målrettet WES til at påvise en genetisk årsag i næsten 30 % af tilfældene (50). Ikke overraskende blev recessive mutationer hyppigere fundet blandt nyfødte og i tilfælde af medfødt sygdom, mens dominerende tilstande normalt manifesterede sig senere i livet. Disse data tyder på, at genetisk nyrestenssygdom er en underdiagnosticeret tilstand på trods af, at den molekylære diagnose potentielt vil påvirke prognose, profylakse og/eller behandling. En begrænsning, der er værd at nævne, er dog en potentiel selektionsbias som følge af rekruttering fra specialiserede nyrestensklinikker i alle tre ovennævnte undersøgelser.

Figur 2. Mutationsanalyse af 30 kendte monogene gener for nefrolithiasis (NL)/nefrocalcinose (NC) hos 268 patienter med NL/NC. (A) Fraktion af monogene årsager i pædiatrisk og voksen subkohorte. (B) Antal monogene årsager på tværs af generne (rødt angiver voksne; orange angiver pædiatriske patienter). Bemærkelsesværdigt er, at SLC7A9 blev fundet hyppigst muteret, især hos unge voksne.
Identificering af nye menneskelige sygdomsgener ved hjælp af kandidatgenmetoden
Mutationsanalyser med højt gennemløb anvendes også til at screene for patogene varianter i forskellige kandidatgener. Et af de mest interessante nylige resultater var opdagelsen af humane mutationer i SLC26A1 (32). Siden den første beskrivelse af Ca-oxalat (CaOx) nyrestensdannelse og NC i Slc26a1 (Sat1)-knockout-mus af Dawson et al. i 2010 har SLC26A1 været et bona fide NL-kandidatgen (51). SLC26A1 koder for en anionbytter, der udtrykkes ved den basolaterale membran i proximale nyretubuli, ileum og jejunum. Ved hjælp af en kandidat-gen-tilgang blev der derfor identificeret patogene varianter hos mennesker med en historie af tidligt indsættende CaOx-NL, nemlig to ikke-relaterede personer med bialleliske missense-varianter (32). Funktionelt blev patogeniciteten af de identificerede varianter påvist in vitro, hvilket førte til intracellulær fejltrafikering og nedsat transportaktivitet (32). Defekt SLC26A1 udgør derfor en ny årsag til CaOx-NL og bør overvejes, når man tester personer for årsager til tilbagevendende CaOx-sten-dannelse.
Nyt klinisk patientregister for arvelig nyrestenssygdom
De fleste epidemiologiske data om stigende prævalens i vestlige lande stammer fra amerikanske databaser. Selv om der er et presserende behov for centraliserede europæiske databaser, er de ikke tilgængelige på nuværende tidspunkt. Da ovennævnte genetiske undersøgelser af prævalensen af arvelig nyrestenssygdom blev udført med små kohorter fra specialiserede centre i både Europa og USA, er en oversættelse til den generelle situation i Europa ikke gyldig. Mens Rare Kidney Stone Consortium i USA udgør en platform, der integrerer og koordinerer register-, grundvidenskabelige og kliniske forskningsaktiviteter for sjældne sygdomme som cystinuri, PH, APRT-mangel, Dent- og Lowe-sygdom, er der i dag hverken i Europa eller i Tyskland gennemført nogen tilsvarende dataindsamling om patienter med arvelig nyrestenssygdom. I samarbejde med det eksisterende europæiske PH-register OxalEurope (professor Bernd Hoppe, universitetet i Bonn) og med støtte fra Deutsche Forschungsgemeinschaft og Else Kröner-Fresenius Stiftung har vi for nylig etableret et klinisk patientregister for arvelig nyrestenssygdom på universitetet i Leipzig. Registret støttes på nationalt plan af de tyske selskaber for nefrologi for voksne (DGfN) og børnenefrologi (GPN). Det er desuden registreret i det tyske register for kliniske forsøg (DRKS-ID: DRKS00012891). Som en grundlæggende del af rekrutteringen til undersøgelsen tilbydes højhastigheds-mutationsanalyse for kendte og nye nyrestengener på forskningsbasis til patienter uden en etableret molekylær diagnose, men med et klinisk billede, der peger på en underliggende genetisk modtagelighed: f.eks. tidlig debutalder (<40 år), positiv familiehistorie, indikative fænotyper som NC, cystinuri eller RTA og alvorligt tilbagevendende NL (>3×) (tabel 2). Mens patienter med en allerede etableret genetisk diagnose generelt indskrives, indgår tilfælde med sekundære NL/NC-årsager, såsom malignitet, sarkoidose og primær hyperparathyroidisme, ikke i den genetiske analyse. For aktivt at indskrive patienter skal et klinisk center normalt have godkendelse fra det lokale Institutional Review Board; en proces, som vi tilbyder vores hjælp og bistand til ved at stille respektive skabeloner til rådighed. Efter etisk godkendelse kan samtykkeerklæringen og de kliniske datablade (f.eks. klinisk spørgeskema) downloades fra vores registerwebsted (http://www.mks-registry.net). For at sikre en grundig klinisk fænotyping vil vi bede om væsentlige patientoplysninger såsom etnicitet, slægtskab, familiehistorie, alder ved debut, recidiv (defineret som hver formodet ny nyrestenbegivenhed), dagligt væskeindtag, kirurgiske indgreb og extrarenal involvering blandt andet. Dokumenterne kan udfyldes af patienten med hjælp fra den tilmeldende læge. Desuden vil biokemiske serumparametre, herunder kreatinin, eGFR, PTH, D-vitamin, elektrolytter, urinsyre og urinanalyse (pH, calcium, fosfat, magnesium, urinsyre, citrat, oxalat og cystin, fortrinsvis fra 24-timers urin, hvis ikke spoturin), samt data om analyse af stensammensætning blive anmodet om ved indskrivningen. I betragtning af at 24-timers urinindsamling og analyse af stensammensætningen ikke udføres rutinemæssigt på alle institutioner, medtager vi disse parametre, når de er tilgængelige. 2-årige kliniske opfølgningsbesøg af de tilmeldte patienter er ønskelige, men ikke obligatoriske. Efter registrering vil de rekrutterende kliniske centre få et personligt login til at indtaste patientdata via vores registerwebsted (http://www.mks-registry.net). Alternativt tilbyder vi at indtaste dataene elektronisk, når de sendes til os på papir. De indtastede data vil blive lagret på en sikret server og kan tilgås af de deltagende kliniske centre for at se deres egne patientdata. På følgende websteder findes yderligere oplysninger:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
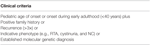
Tabel 2. Inklusionskriterier for mutationsanalyse i klinisk patientregister.
Sammenfattende er nyrestenssygdom en stadig mere udbredt tilstand, som er klinisk heterogen og dårligt forstået, navnlig dens genetiske drivkræfter. Som en række nyere undersøgelser viste, er monogene tilstande højst sandsynligt undervurderet i prævalens. Ved at oprette et centraliseret patientregister over arvelig nyrestenssygdom vil vi bidrage til, i det mindste delvist, at afhjælpe den store mangel på viden om genetisk betinget nyrestenssygdom. I denne forbindelse er kliniske registre værdifulde kilder af flere grunde: For det første vil det være afgørende for en mere præcis stratificering af patienterne i fremtidige kliniske forskningsundersøgelser, at der kan udarbejdes bedre sammenhænge mellem fænotype og genotype. For det andet vil identifikation af nye sygdomsgener med nye sygdomsmekanismer mindske hullet i den ukendte NL/NC-ætiologi, og for det tredje vil dechifreringen af nye molekylære mål bidrage til at bane vejen for udvikling af lægemidler til forebyggelse af tilbagefald i alvorligt ramte familier.
Ethisk erklæring
Denne undersøgelse blev gennemført i overensstemmelse med anbefalingerne fra “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” med skriftligt informeret samtykke fra alle forsøgspersoner. Alle forsøgspersoner gav skriftligt informeret samtykke i overensstemmelse med Helsinki-erklæringen. Protokollen blev godkendt af “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig.”
Author Contributions
JH udtænkte og skrev manuskriptet. BH, AS, LM og RS redigerede manuskriptet og opbyggede registrets infrastruktur, der præsenteres for læseren.
Interessekonflikter
Forfatterne erklærer, at forskningen blev udført uden kommercielle eller økonomiske relationer, der kunne opfattes som en potentiel interessekonflikt.
Funding
Forskningen er finansieret af projekttilskud fra DFG (HA 6908/2-1) og EKFS (2016_A52) til JH. Dette arbejde blev yderligere støttet af forbundsministeriet for uddannelse og forskning (BMBF), Tyskland, FKZ: 01EO1501 til JH.
1. Scales CD Jr, Smith AC, Hanley JM, Hanley JM, Saigal CS. Prævalens af nyresten i USA. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Genetik af hypercalciurisk nefrolithiasis: renal stensygdom. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Fremskridt i forståelsen af genetikken af calciumholdig nefrolithiasis. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sekvensvarianter i CLDN14-genet associeres med nyresten og knoglemineraltæthed. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-rolle of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Almindelige og sjældne varianter associeret med nyresten og biokemiske træk. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 påvirker urinsyrekoncentrationer med udpræget kønsspecifikke virkninger. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutationer i glukosetransportør 9-genet SLC2A9 forårsager renal hypouricæmi. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identifikation og karakterisering af et gen med basesubstitutioner, der er forbundet med den absorptive hypercalciuria-fænotype og lav spinal knogletæthed. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting af peroxisomal L-alanin:glyoxylataminotransferase til mitokondrier hos patienter med primær hyperoxaluri afhænger af aktivering af en kryptisk mitokondriel targeting-sekvens ved hjælp af en punktmutation. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Human adeninphosphoribosyltransferase. Identifikation af alleliske mutationer på nukleotidniveau som årsag til fuldstændig mangel på enzymet. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutationer i ATP6N1B, der koder for en ny ny nyrevakuolær vacuolar protonpumpe 116-kD-underenhed, forårsager recessiv distal renal tubulær acidose med bevaret hørelse. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutationer i det gen, der koder for B1-underenheden af H+-ATPase, forårsager renal tubulær acidose med sensorineural døvhed. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II-mangelsyndrom i en belgisk familie er forårsaget af en punktmutation på en invariant histidinrest (107 His→Tyr): komplet struktur af det normale menneskelige CA II-gen. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. Et familiært syndrom af hypokalcæmi med hypercalciuri som følge af mutationer i den kalciumsensoriske receptor. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. Et fælles molekylært grundlag for tre arvelige nyrestenssygdomme. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutationer i kloridkanalgenet, CLCNKB, forårsager Bartters syndrom type III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, et renal tight junction-protein, der er nødvendigt for paracellulær Mg2+-resorption. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutationer i tight-junction-genet claudin 19 (CLDN19) er forbundet med renal magnesiumsvind, nyresvigt og alvorlig okulær involvering. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutationer i CYP24A1 og idiopatisk infantil hypercalcæmi hos børn. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nefrocalcinose (emaljenyresyndrom) forårsaget af autosomalt recessive FAM20A-mutationer. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Genet, der koder for hydroxypyruvatreduktase (GRHPR), er muteret hos patienter med primær hyperoxaluria type II. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. HNF4A R76W-mutationen forårsager atypisk dominant Fanconis syndrom ud over en β-cellefænotype. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutationer i DHDPSL er ansvarlige for primær hyperoxaluria type III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identifikation af 17 uafhængige mutationer, der er ansvarlige for hypoxanthin-guaninphosphoribosyltransferase (HPRT)-mangel hos mennesker. Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetisk heterogenitet af Bartters syndrom afsløret af mutationer i K+-kanalen, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartter’s syndrom og MAGED2-mutationer. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Tightly linked flanking markers for Lowe oculocerebrorenal syndrom, med anvendelse til bærervurdering. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartters syndrom, hypokalaemisk alkalose med hypercalciuri, er forårsaget af mutationer i Na-K-2Cl-kotransporter NKCC2. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutationer i SLC26A1 forårsager nefrolithiasis. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molekylær identifikation af en renal urat-anionudveksler, der regulerer bloduratniveauet. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditær hypofosfatematisk rakitis med hyperkalciuri skyldes mutationer i natrium-fosfat cotransporter-genet SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinuria forårsaget af mutationer i rBAT, et gen, der er involveret i transport af cystin. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familiær distal renal tubulær acidose er associeret med mutationer i genet for anionbyttere i røde blodlegemer (Band 3, AE1). J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Non-type I cystinuria forårsaget af mutationer i SLC7A9, der koder for en underenhed (bo,+AT) af rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1-mutationer og respons på renalt parathyreoideahormon. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Suggestive beviser for et modtagelighedsgen nær D-vitaminreceptorlokus i idiopatisk calciumstensdannelse. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identifikation af to mutationer i det humane xanthindehydrogenase-gen, der er ansvarlig for klassisk type I xanthinuria. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Hildebrandt F. Genetiske nyresygdomme. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutationer i SLC34A3/NPT2c er forbundet med nyresten og nefrocalcinose. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Kliniske og biokemiske fænotyper hos voksne med monoalleliske og bialleliske CYP24A1-mutationer: bevis for gen-doseeffekt. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Récents acquis diagnostiques et thérapeutiques. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. Højhastighedsmutationsanalyse i patienter med en nephronophthisis-associeret ciliopati ved anvendelse af multiplexed barcoded array-baseret PCR-amplifikation og næste generations sekventering. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identifikation af 99 nye mutationer i en verdensomspændende kohorte på 1 056 patienter med en nefronofte-relateret ciliopati. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. 14 monogene gener tegner sig for 15 % af nefrolithiasis/nefrocalcinose. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prævalens af monogene årsager hos pædiatriske patienter med nefrolithiasis eller nefrocalcinose. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolithiasis og hepatotoksicitet er knyttet til aniontransportøren Sat1 hos mus. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar