Alexei Drummond, Andrew Rambaut, Remco Bouckaert og Walter Xie
1 Introduktion
Denne vejledning introducerer BEAST-softwaren til bayesiansk evolutionær analyse ved hjælp af en simpel vejledning. Vejledningen involverer co-estimation af en gen-fylogeni og tilhørende divergenstider i tilstedeværelsen af kalibreringsinformation fra fossile beviser.
Du skal have følgende software til din rådighed:
- BEAST – denne pakke indeholder programmet BEAST, BEAUti, TreeAnnotator og andre hjælpeprogrammer. Denne vejledning er skrevet til BEAST v2.2.x, som har understøttelse for flere partitioner. Den kan downloades fra http://www.beast2.org/.
- Tracer – dette program bruges til at udforske output fra BEAST (og andre Bayesian MCMC-programmer). Det opsummerer grafisk og kvantitativt fordelingerne af kontinuerte parametre og giver diagnostiske oplysninger. I skrivende stund er den aktuelle version v1.6. Den kan downloades fra
http://tree.bio.ed.ac.uk/software/. - FigTree – dette er et program til visning og udskrivning af molekylære fylogenier, især dem, der er opnået ved hjælp afBEAST. I skrivende stund er den aktuelle version v1.4.2. Det kan downloades fra http://tree.bio.ed.ac.uk/software/.
Denne vejledning vil guide dig gennem analysen af en alignment af sekvenser udtaget fra tolv primatarter (se figur 1). Målet er at estimere fylogenien, udviklingshastigheden for hver enkelt slægt og alderen for de ukalibreredeancestrale divergenser.
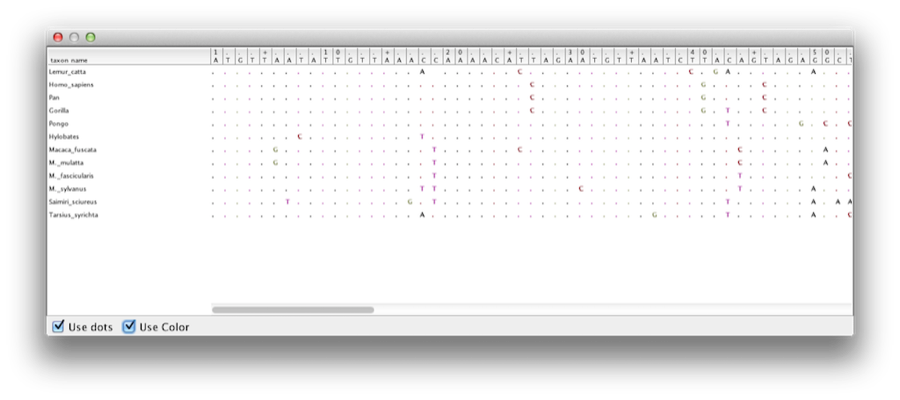
Figur 1: Del af alignmentet for primater.
Det første skridt vil være at konvertere en NEXUS-fil med en DATA- eller CHARACTERS-blok til en BEAST XML-inputfil. Dette gøres ved hjælp af programmet BEAUti (som står for Bayesian Evolutionary Analysis Utility). Dette er et brugervenligt program til at indstille den evolutionære model og indstillingerne for MCMC-analysen. Det andet trin er faktisk at køre BEAST ved hjælp af den inputfil, der er genereret af BEAUTi, og som indeholder data, model og analyseindstillinger. Det sidste trin er at udforske resultatet af BEAST for at diagnosticere problemer og sammenfatte resultaterne.
2 BEAUti
Programmet BEAUti er et brugervenligt program til indstilling af modelparametre til BEAST. Kør BEAUti ved at dobbeltklikke på dets ikon. Når BEAUti er kørt, vil det se ens ud, uanset hvilket computersystem det kører på. I denne vejledning anvendes Mac OS X-versionen i figurerne, men Linux- og Windows-versionerne vil have samme layout og funktionalitet.
2.1 Indlæsning af NEXUS-filen
For at indlæse en justering i NEXUS-format skal du blot vælge Import Alignment… i menuen File (Fil) eller trække filen ind i midten af Partitions-panelet.
Eksempelfilen kaldet primate-mtDNA.nex er tilgængelig fra mappen examples/nexus/ til Mac og Linux og examples/nexus/til Windows inde i den mappe, hvor BEAST blev installeret. denne fil indeholder en alignment af sekvenser af 12 arter af primater.
Et vindue Add Partition (Figur 2) vil dukke op, hvis den relaterede pakke er installeret. Hvis du bruger “ren” BEAST 2, kan du gå videre til næste afsnit. Ellers skal du vælge Add Alignment og klikke på OK for at fortsætte.
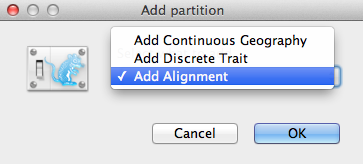
Figur 2: Vinduet Add Partition (vises kun, hvis relaterede pakker er installeret).
Hvis der er kodningsoverlapninger i partitionerne, vises vinduet med en advarselsmeddelelse (Figur 3). Læs og klik på OK for at fortsætte.
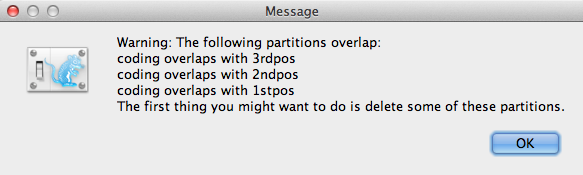
Figur 3: Vindue med advarselsmeddelelse (vises kun, hvis der er kodningsoverlapninger i partitioner).
Når de er indlæst, vises fem karakterpartitioner i hovedpanelet (Figur 4). Justeringen er opdelt i en proteinkodende del og en ikke-kodende del,og den kodende del er opdelt i kodonpositioner 1, 2 og 3. Du skal fjerne partitionen “coding”, før du fortsætter til næste trin, da den henviser til de samme nukleotider som partitionerne “1stpos”, “2ndpos” og “3rdpos”. For at fjerne partitionen “kodning” vælges rækken, og der klikkes på knappen “-” nederst i tabellen. Du kan se alignmentet ved at dobbeltklikke på partitionen.
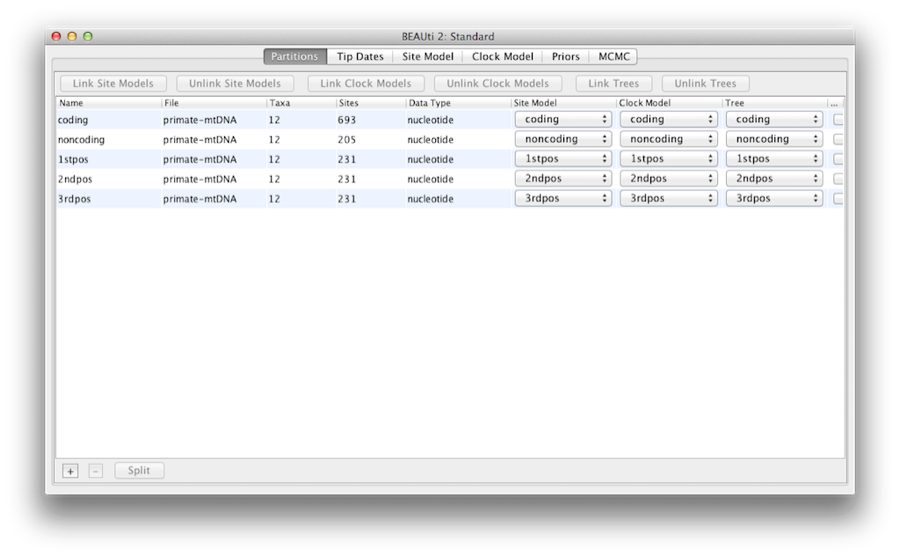
Figur 4: Et skærmbillede af fanen data i BEAUti. Dette og alle følgende skærmbilleder blev taget på en Apple-computer med Mac OS X og vil se lidt anderledes ud på andre styresystemer.
Link/Unlink partition models
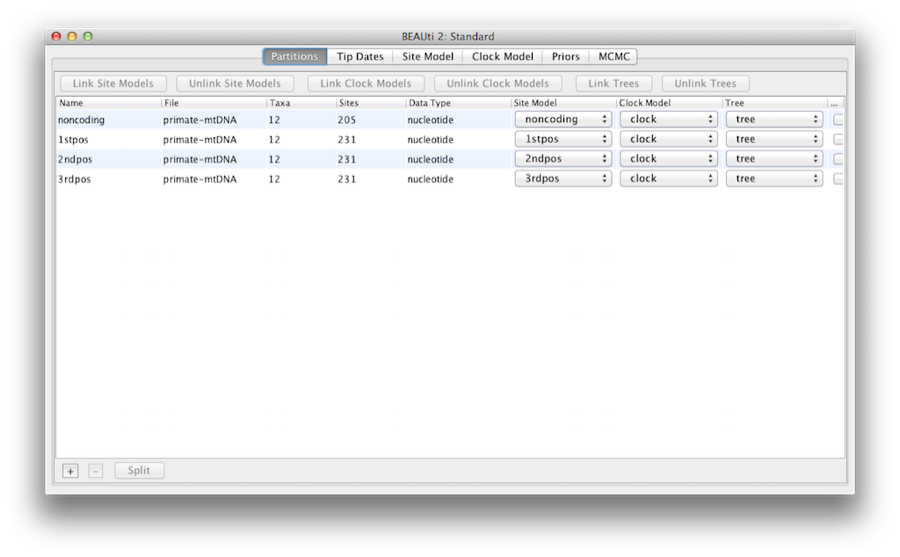
Figur 5: Et skærmbillede af fanen Partitionsfanen i BEAUti efter at have linket og omdøbt urmodellen og træet.
Da sekvenserne er forbundet (dvs. de er alle fra det mitokondrielle genom, som ikke menes at undergå rekombination hos fugle og pattedyr), har de samme forfædre, så partitionerne bør dele samme tidstræ i modellen. For enkelhedens skyld antager vi også, at partitionerne har den samme udviklingshastighed for hver gren og dermed den samme “urmodel”. vi begrænser vores modellering af hastighedsheterogenitet til at omfatte heterogenitet mellem steder inden for hver partition og tillader også, at partitionerne har forskellige gennemsnitlige udviklingshastigheder.
Så på dette tidspunkt bliver vi nødt til at sammenkæde urmodellen og træet. I panelet Partitions skal du vælge alle fire partitioner i tabellen (eller ingen, som standard er alle partitioner berørt) og klikke på knappen Link Trees og derefter på knappen Link Clock Models (se figur 5). Klik derefter på den første rullemenu i kolonnen Clock Model (Urmodel), og omdøb den delte urmodel til “clock” (ur). Omdøb på samme måde det delte træ til “tree”. Dette vil gøre følgende indstillinger og genererede logfiler lettere at læse.
2.2 Indstilling af substitutionsmodellen
Det næste trin er at opsætte substitutionsmodellen. Vælg derefter fanen Site Models-fanen øverst i hovedvinduet (vi springer fanen Tip Dates over, da alle taxa er fra samtidige prøver). Dette vil afsløre indstillingerne for den evolutionære model for BEAST. De tilgængelige indstillinger afhænger af, om dataene er nukleotider eller aminosyrer, binære data eller generelle data. De indstillinger, der vises efter indlæsning af primatnukleotid-alligeringen, vil være standardværdierne for nukleotiddata, så vi skal foretage nogle ændringer.
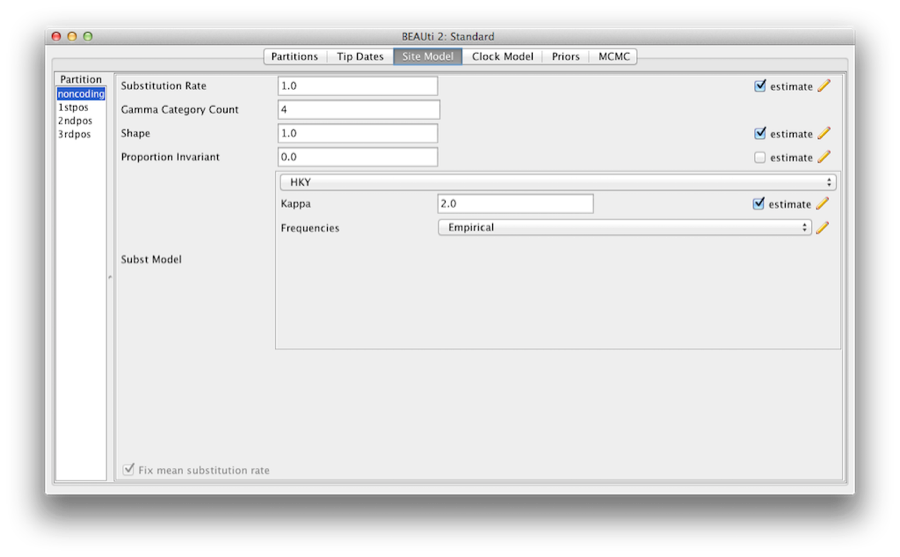
Figur 6: Et skærmbillede af fanen site model i BEAUti.
De fleste af modellerne burde være velkendte for dig. Først skal du indstille Gamma Category Count til 4 og derefter afkrydse feltet “estimate” for Shape-parameteren. Dette vil gøre det muligt at modellere hastighedsvariationen mellem steder i hver partition. Bemærk, at 4 til 6 kategorier fungerer tilstrækkeligt godt for de fleste datasæt, mens det tager mere tid at beregne flere kategorier for en lille ekstra fordel at have flere kategorier. Vi lader posten Proportion Invariant være indstillet til nul.
Dernæst vælges HKY fra drop-down-menuen Subst Model. Ideelt set bør der vælges en substitutionsmodel, der passer bedst til dataene for hver partition, men her bruger vi for enkelhedens skyld HKY for alle partitioner. Vælg endvidere Empirisk i rullemenuen Frekvenser. Dette vil fastsætte frekvenserne til de proportioner, der er observeret i dataene (for hver partition individuelt, når vi har fjernet forbindelsen mellem stedmodellerne). Denne fremgangsmåde betyder, at vi kan få en god tilpasning til dataene uden eksplicit estimering af disse parametre. Vi gør det her blot for at gøre logfilerne lidt kortere og mere læsevenlige i senere dele af øvelsen.
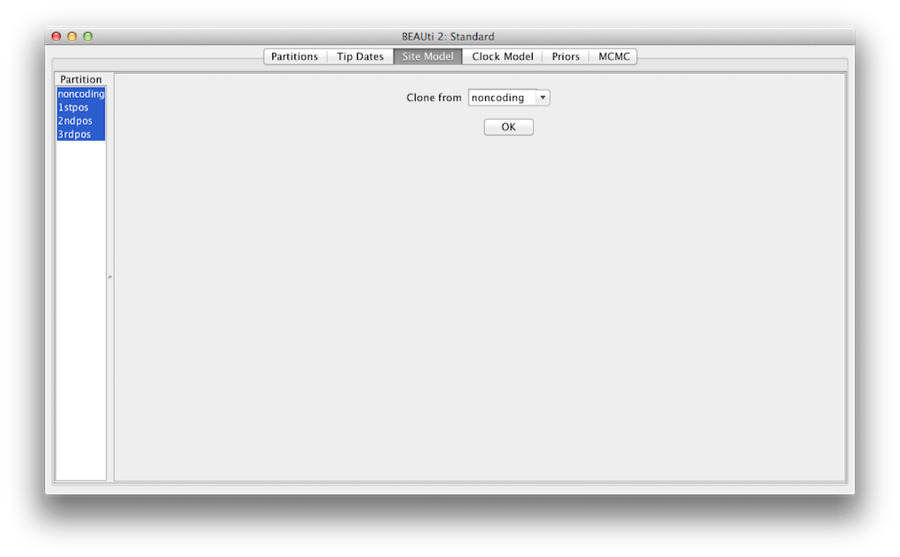
Figur 7: Klonkonfiguration fra en lokalitetsmodel til andre.
Finalt markeres afkrydsningsfeltet “estimat” for parameteren Substitutionsrate, og afkrydsningsfeltet Fix mean mutation rate markeres. Dette vil gøre det muligt for de enkelte partitioner at få deres relative hastigheder estimeret for de ikke-forbundne stedmodeller (figur 6).
Sidst skal du holde ‘shift’-tasten nede for at vælge alle stedmodeller i venstre side, og klikke på OK for at klone indstillingen fra noncoding til 1stpos, 2ndpos og 3rdpos (figur 7). Gennemgå hver enkelt stedmodel, og som du kan se, er deres konfigurationer nu ens.
2.3 Indstilling af urmodellen
Det næste trin er at vælge fanen Clock Models (Urmodeller) øverst i hovedvinduet. Det er her, vi vælger den molekylære urmodel. I denne øvelse lader vi valget stå på standardværdien af et strengt molekylært ur, fordi disse data er meget urlignende og ikke har brug for hastighedsvariation mellem grene for at blive inkluderet i modellen.
For at teste, om der er tale om et ur, kan du (i) køre analysen med en afslappet urmodel og kontrollere, hvor stor variation mellem hastighederne der er impliceret af dataene (se variationskoefficient for mere om dette), eller(ii) udføre en modelsammenligning mellem et strengt og afslappet ur ved hjælp af pathsampling, eller (iii) bruge en tilfældig lokal urmodel, som eksplicit overvejer, om hver gren i træet har brug for sin egen grenhastighed.
2.4 Priors
Fanen Priors giver mulighed for at angive priors for hver parameter i themodellen. De modelvalg, der er foretaget i fanerne Site model og Clock model, resulterer i, at der indgår forskellige parametre i modellen, og disse vises i fanen Priors (se figur 8).
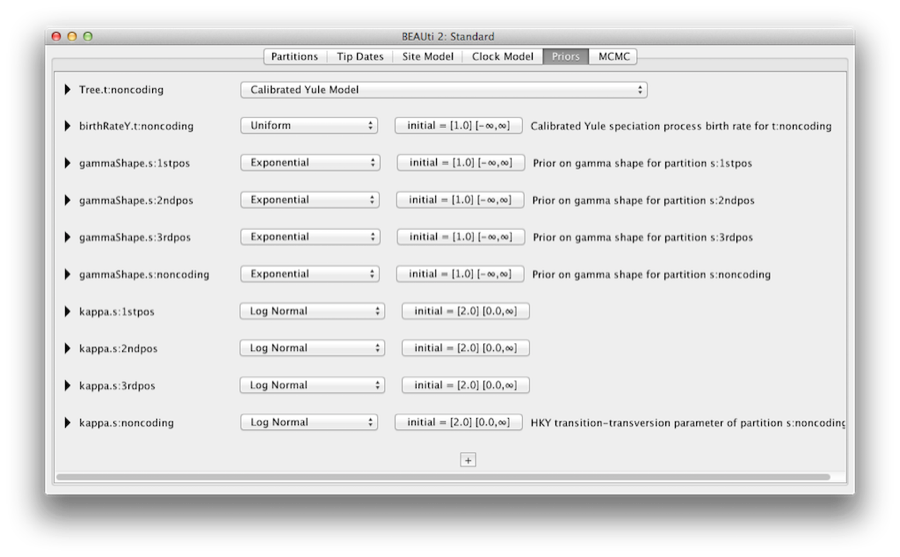
Figur 8: Et skærmbillede af fanen Priors i BEAUti.
Her angiver vi også, at vi ønsker at bruge den kalibrerede Yule-model som træprioritet. Yule-modellen er en simpel model for artsdannelse, som generelt er mere hensigtsmæssig, når der tages hensyn til sekvenser fra forskellige arter. Vælg Calibrated Yule Model i dropdown-menuen Tree prior.
2.4.4.1 Definition af kalibreringsnoden
Vi skal nu angive en prioritetsfordeling på den kalibrerede node, baseret på voresfossilviden. Dette er kendt som kalibrering af vores træ. Hvis du vil definere en ekstra prioritet, skal du trykke på den lille +-knap under listen over prioriteter. Hvis den ikke er synlig i din visning, skal du scrolle ned i panelet til bunden for at finde +-knappen. Du vil se en dialogboks, der giver dig mulighed for at definere en delmængde af taxaerne i det fylogenetiske træ. Når du har oprettet et taxasæt, vil du senere kunne tilføje kalibreringsoplysninger for dets seneste fællesancestor (MRCA).
Nævn taxa-sættet ved at udfylde angivelsen for taxa-sættets etiket. Kald det human-chimpanse, da det vil indeholde taxaerne for Homo sapiens og Pan. I listen nedenfor kan du se de tilgængelige taxa. Vælg hver af de to taxa på skift, og tryk på > >-pilknappen. (Figur 9) Klik på OK, og det nydefinerede taxasæt vil blive tilføjet i prior-listen. da dette er en kalibreret knude, der skal bruges sammen med den kalibrerede Yule-prior, skal monofylien håndhæves, så marker afkrydsningsfeltet Monophyletic. Dette vil begrænse træets topologi, så grupperingen menneske-chimpanse holdes monofyletisk i løbet af MCMC-analysen.
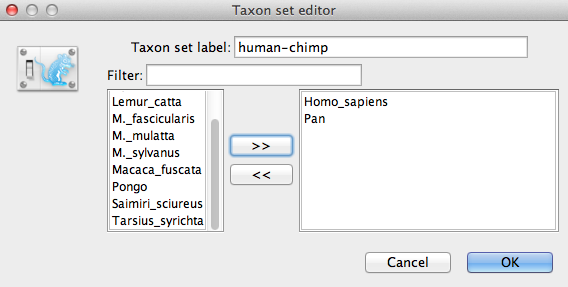
Figur 9: Taxonsæt-editor i BEAUti.
For at kode kalibreringsoplysningerne skal vi angive en fordeling for MRCA for human-chimpanse. vælg Log-normalfordelingen fra drop-down-menuen til højre for den nyligt tilføjede human-chimpanse.prior. Klik på den sorte trekant, og der vises en graf over sandsynlighedsdensitetsfunktionen sammen med parametre for lognormalfordelingen. vi indstiller M=1,78 og S=0,085, hvilket vil angive en fordeling centreret omkring 6 millioner år med en standardafvigelse på ca. 0,5 millioner år. Dette vil give et centralt 95% sandsynlighedsområde, der dækker 5-7 Mya. Dette svarer nogenlunde til det nuværende konsensusstimat af datoen for den seneste fælles forfader for mennesker og chimpanser (Figur 10).
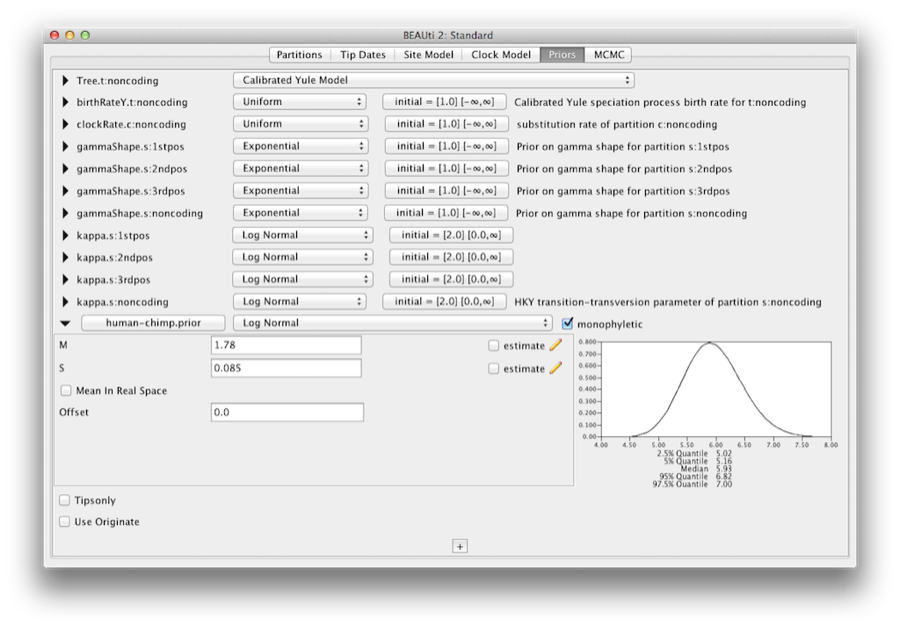
Figur 10: Et skærmbillede af indstillingerne for kalibreringsprioritet i panelet Priors i BEAUti.
Vi bør overbevise os selv om, at de priorer, der vises i priors-panelet, virkelig afspejler den forudgående information, vi har om modellens parametre. Endelig vil vi også angive nogle diffuse “uinformative”, men korrekte priorer på den overordnede molekylære urhastighed (clockRate) og artsdannelseshastigheden (birthRateY) i Yule-træets prioriteter. For hver af dem skal du vælge Gamma fra rullemenuen og ved hjælp af pileknappen udvide visningen for at afsløre parametrene for Gamma-prioriteten. For både clock rate og Yule birth rate indstilles parameteren Alpha(shape) til 0,001 og parameteren Beta(scale) til 1000 (figur 11).
Som standard har hver af gammaformparametrene en eksponentiel prioritetsfordeling med en middelværdi på 1. Dette indebærer (se figur 3.7), at vi forventer en mindre variation. Som standard har kappa-parametrene for HKY-modellen en lognormal(1,1,25)-prioritetsfordeling, hvilket i det store og hele stemmer overens med empirisk dokumentation for intervallet af realistiske værdier for overgangs-/transversionsbias. Disse standardprioriteter bevares, da de er velegnede til denne særlige analyse.
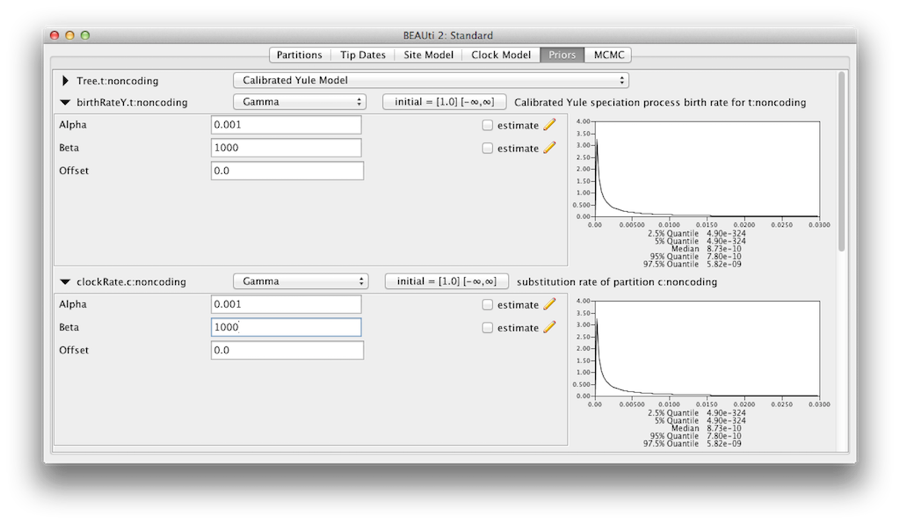
Figur 11: Gammaprioritet.
2.5 Indstilling af MCMC-indstillinger
Den næste fane, MCMC, indeholder mere generelle indstillinger til kontrol af længden af MCMC-kørslen og filnavnene.
Først har vi Chain Length (kædelængde). Dette er det antal trin, som MCMC’en vil foretage i kæden, før den afsluttes. Hvor lang denne skal være, afhænger af datasættets størrelse, modellens kompleksitet og kvaliteten af det ønskede svar. Standardværdien på 10.000.000 er helt vilkårlig og bør justeres i forhold til størrelsen af dit datasæt. Lad os for dette datasæt sætte kædelængden til 6.000.000.000, da dette vil køre rimeligt hurtigt på de fleste moderne computere (et par minutter).
Feltet Store Every bestemmer, hvor ofte tilstanden skal gemmes i en fil. Det er nyttigt at gemme tilstanden periodisk i situationer, hvor computermiljøet ikke er særlig pålideligt, og hvor en BEAST-kørsel kan afbrydes. Når du har en gemt kopi af den seneste tilstand, kan du genoptage kæden i stedet for at starte forfra, så du ikke behøver at gennemgå burn-in igen.Feltet Pre Burnin angiver antallet af prøver, der ikke logges helt fra begyndelsen af analysen. Vi lader felterne Store Every og Pre Burnin være indstillet til deres standardværdier. Nedenfor er der oplysninger om logfilerne. Hver enkelt kan udvides ved at klikke på den sorte trekant.
De næste indstillinger angiver, hvor ofte parameterværdierne i Markovkæden skal vises på skærmen og registreres i logfilen.Skærmudgangen er blot til overvågning af programmets fremskridt og kan indstilles til en hvilken som helst værdi (selvom hvis den er indstillet for lille, vil selve mængden af oplysninger, der vises på skærmen, faktisk bremse programmet). For logfilen skal værdien indstilles i forhold til kædens samlede længde. En for hyppig prøveudtagning vil resultere i meget store filer med ringe ekstra fordele med hensyn til analysens nøjagtighed. Hvis der tages for sjældent prøver, vil logfilen ikke registrere tilstrækkelige oplysninger om parametrenes fordeling. Du vil sandsynligvis tilstræbe ikke at gemme mere end 10.000 prøver, så dette bør ikke være mindre end kædelængde / 10.000.
I denne øvelse indstiller vi trace log- og tree log-frekvensen til 1.000 og screen log-frekvensen til 10.000. Angiv også Primates.log som filnavn for trace log-filen og Primates.trees som filnavn for tree log-filen.Sørg for, at filnavnefeltet for skærmlogfilen er tomt, ellers vil skærmlogfilen ikke blive skrevet til skærmen.
- Hvis du bruger Windows-operativsystemet, foreslår vi, at du tilføjer suffikset .txt til begge disse (altså Primates.log.txt og Primates.trees.txt), så Windows kan genkende dem som tekstfiler.
2.6 Generering af BEAST XML-filen
Vi er nu klar til at oprette BEAST XML-filen. For at gøre dette skal du vælge indstillingen Save (Gem) i menuen File (Fil). Kontroller standardprioriteterne, og gem filen med et passende navn (vi plejer at afslutte filnavnet med .xml, dvs. Primates.xml).Vi er nu klar til at køre filen gennem BEAST.
3 Kørsel af BEAST
Figur 12: Et skærmbillede af BEAST.
Kør nu BEAST, og når den beder om en inputfil, skal du angive din nyligt oprettede XML-fil som input. BEAST vil derefter køre, indtil den er færdig med at rapportere oplysninger til skærmen. De faktiske resultatfiler gemmes på disken på samme sted som din inputfil. Output til skærmen vil se nogenlunde sådan her ud:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Bemærk, at der i starten er nogle nyttige oplysninger om tilpasningerne, og hvilke træ-sandsynligheder der er anvendt. Desuden er alle citater, der er relevante for analysen, nævnt i starten af kørslen, hvilket let kan kopieres tilomanuskripter, der rapporterer om analysen. Derefter følger rapportering af kæden,som giver en vis feedback i realtid om kædens forløb.
I slutningen udskrives en operatøranalyse, som opregner alle operatorer, der er anvendt i analysen, sammen med hvor ofte operatoren blev forsøgt, accepteret og afvist(se henholdsvis kolonnerne #total, #accept og #reject). Acceptprocenten er den andel af gange, hvor en operatør er accepteret, når den er valgt til at lave et forslag. Generelt er en høj acceptprocent, f.eks. over 0,5, et tegn på, at forslagene er konservative og ikke udforsker parameterrummet effektivt. På den anden side viser en lav acceptprocent, at forslagene er for aggressive og næsten altid resulterer i en tilstand, der afvises på grund af dens lave posterior.Både for høje og for lave acceptprocenter resulterer i lave ESS-værdier. En acceptrate på 0,234 er målet (baseret på meget begrænsede beviser leveret af ) for mange (men ikke alle) operatører, der er implementeret i BEAST.
Nogle operatører har en indstillings parameter, f.eks. skalafaktoren for ascale parameteren. Hvis den endelige acceptprocent ikke er i nærheden af målet, vil BEAST foreslå en ny værdi for indstillings parameteren, som udskrives i operatøranalysen. I dette tilfælde er alle acceptprocenter gode for de operatører, der har indstillings parametre. Operatorer uden indstillings parametre omfatter wideexchange- og Wilson-Balding-operatorer i denne analyse. Begge disse operatorer forsøger at ændre træets topologi med store skridt, men da dataene i overvejende grad støtter en enkelt topologi, afvises disse radikale forslag næsten altid.
4 Analyse af resultaterne
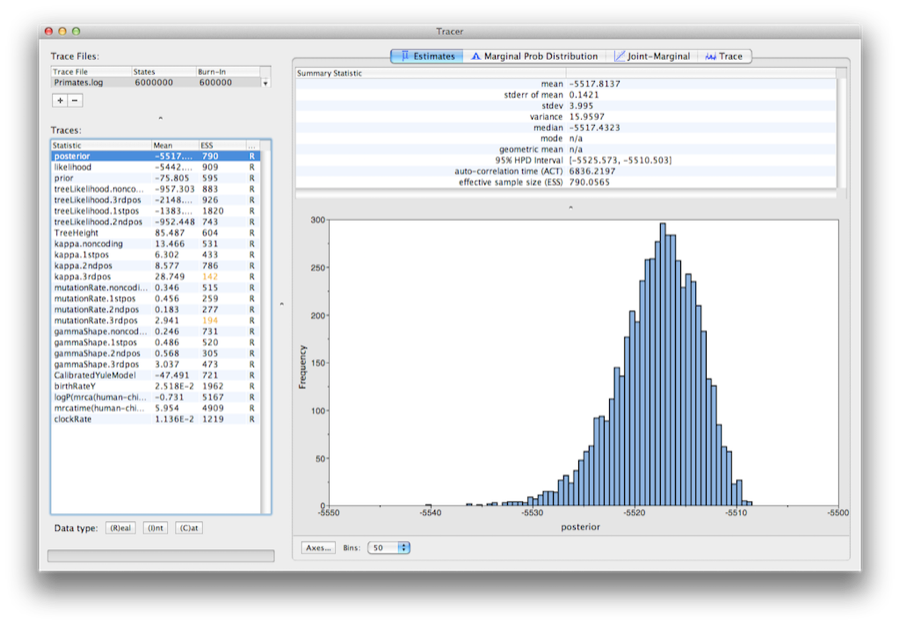
Figur 13: Et skærmbillede af Tracer v1.6.
Kør programmet kaldet Tracer for at analysere output fra BEAST. Når hovedvinduet er åbnet, skal du vælge Import Trace File… i menuen File og vælge den fil, somBEAST har oprettet ved navn Primates.log (Figur 13).
Husk, at MCMC er en stokastisk algoritme, så de faktiske tal vil ikke være nøjagtigt de samme som dem, der er afbildet på figuren.
I venstre side er der en liste over de forskellige størrelser, som BEAST har logget til filen. Der er spor for posterior (dette er den naturlige logaritme af produktet af træets sandsynlighed og den forudgående tæthed) og de kontinuerte parametre. Ved at vælge et spor i venstre side vises analyser for dette spor i højre side, afhængigt af den valgte fane. Ved første åbning er “posterior”-sporet valgt, og forskellige statistikker for dette spor vises under fanen Estimates. øverst til højre i vinduet er der en tabel med beregnede statistikker for det valgte spor.
Vælg parameteren clockRate i den venstre liste for at se på den gennemsnitlige udviklingshastighed (som gennemsnit over hele træet og alle steder). Tracer vil tegne et (marginalt posterior) histogram for den valgte statistik og også give dig en oversigtsstatistik som f.eks. middelværdi og median. 95% HPD står for højeste posterior tæthedsinterval og repræsenterer det mest kompakte interval for den valgte parameter, der indeholder 95% af den posterior sandsynlighed. Det kan løst opfattes som en Bayesiansk analog til et konfidensinterval. TreeHeight-parameteren giver den marginale posteriorfordeling af alderen på roden af hele træet.
Vælg TreeHeight-parameteren, og Ctrl-klik derefter på mrcatime(human-chimp) (Command-klik på Mac OS X). Dette vil vise en visning af rodens alder og den kalibrerings-MRCA, som vi angav tidligere i BEAUti. Du kan verificere, at den divergens, som vi brugte til at kalibrere træet(mrcatime(human-chimp))) har en posteriorfordeling, der passer til den priorfordeling, vi specificerede (Figur 14).
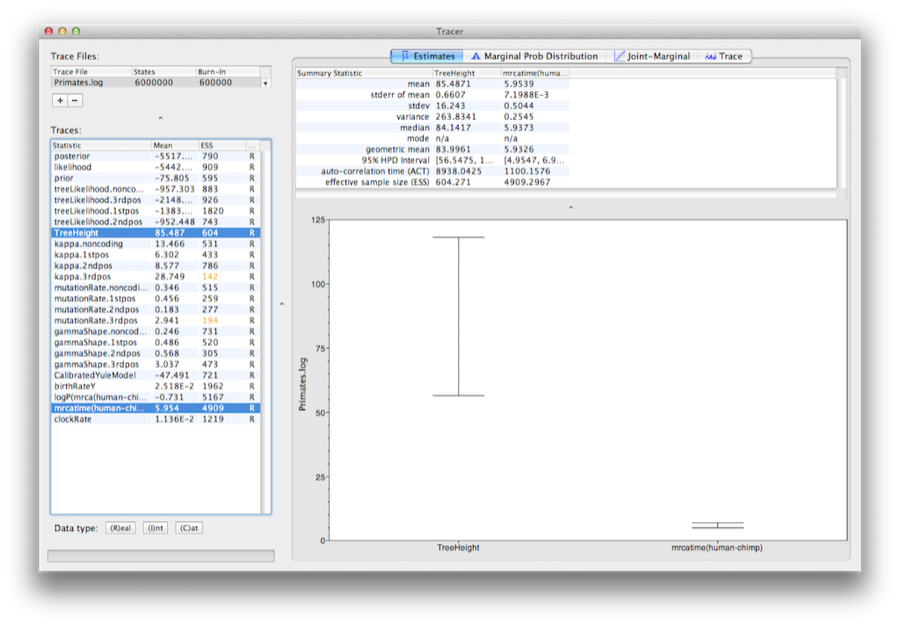
Figur 14: Et skærmbillede af de 95 % HPD-intervaller for rodhøjden og den brugerspecificerede (human-chimp) MRCA i Tracer.
5 Marginale posteriorestimater
For at vise de relative satser for de fire partitioner skal du vælge mutationRate-parameteren for hver af de fire partitioner og vælge fanen marginal tæthed i Tracer.Figur 15 viser marginal tæthederne for de relative substitutionsrater. Plottet viser, at kodonpositioner 1 og 2 har væsentligt forskellige hastigheder (0,456 mod 0,183), og begge er langt langsommere end kodonposition 3 med en relativ hastighed på2,941. Den ikke-kodende partition har en hastighed, der ligger mellem kodonpositioner 1 og 2 (0,346). Samlet set tyder dette resultat på en stærk rensende selektion i både de kodende og ikke-kodende regioner i alignmentet.
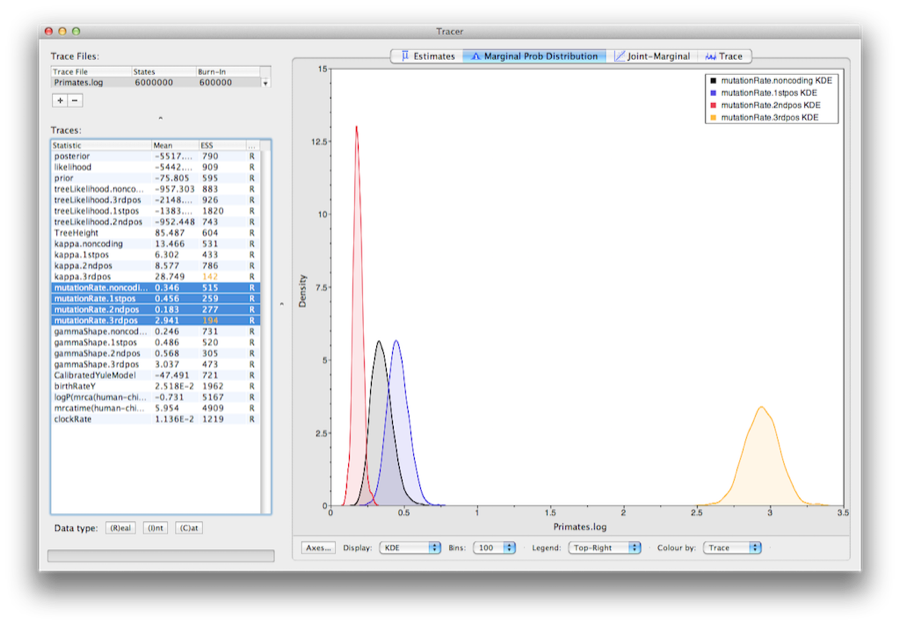
Figur 15: Et skærmbillede af de marginale posterior-tætheder af de relative substitutionshastigheder for de fire partitioner (i forhold til den stedvægtede middelhastighed).
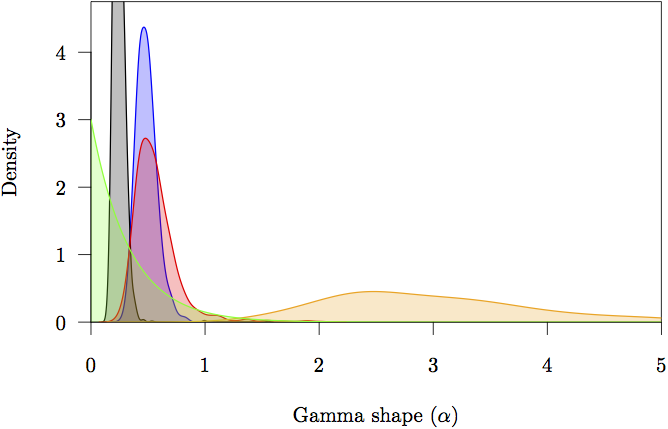
Figur 16: De marginale prior- og posteriortætheder for form (α)-parametrene. Prioriteten er angivet med gråt. Posteriortæthedsskønnet for hver partition er også vist: ikke-kodende (orange) og første (rød), anden (grøn) og tredje (blå) codonpositioner.
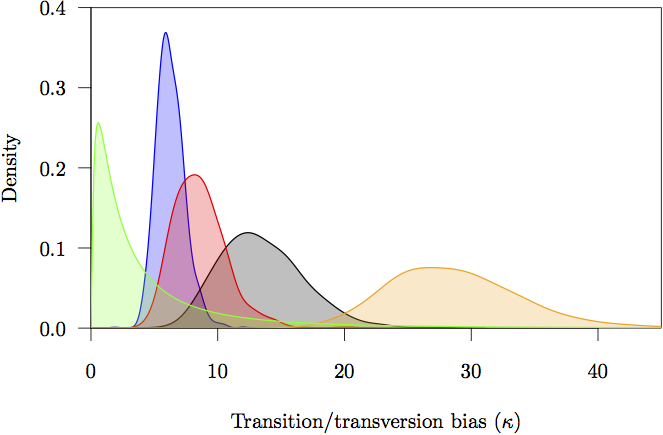
Figur 17: De marginale prior- og posteriortætheder for overgangs-/overgangsbias (κ)-parametrene. Prioriteten er gråtonet. Posteriordensitetsestimatet for hver partition er også vist: ikke-kodende (orange) og første (rød), anden (grøn) og tredje (blå) kodonpositioner.
Spørgsmål
Hvad er den estimerede molekylære udviklingshastighed for dette genetræ (medtag HPD-intervallet på 95 %)?
Hvilke fejlkilder omfatter dette skøn?
Hvor gammel er træets rod (angiv middelværdien og det 95 % HPD-interval)?
6 Opnåelse af et skøn over det fylogenetiske træ
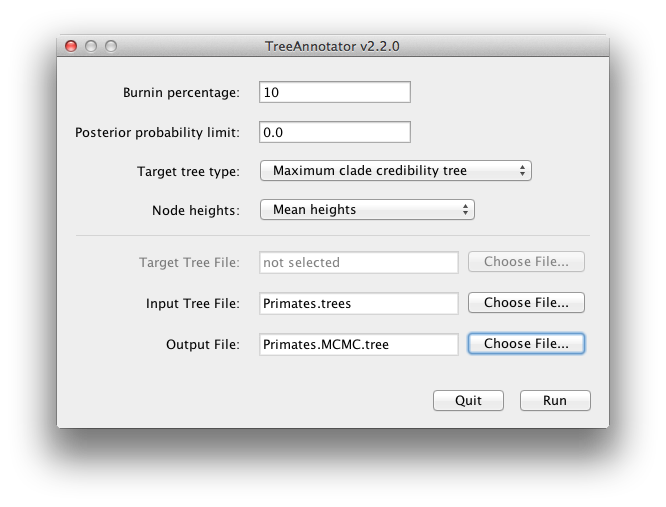
BEAST producerer også en efterfølgende stikprøve af fylogenetiske tidstræer sammen med sin stikprøve af parameterestimater. Disse skal sammenfattes ved hjælp af programmet TreeAnnotator. Dette vil tage mængden af træer og finde det bedst understøttede træ. Det vil derefter annotere dette repræsentative sammenfattende træ med gennemsnitsalderen for alle thenodes og de tilsvarende 95% HPD-intervaller. Det vil også beregne den efterfølgende kladesandsynlighed for hver knude. Kør TreeAnnotator-programmet, og indstil det som vist i figur 18.
Figur 18: Et skærmbillede af TreeAnnotator.
Burnin er antallet af træer, der skal fjernes fra starten af prøven. I modsætning til Tracer, som angiver antallet af trin som burnin, skal du i TreeAnnotator angive det faktiske antal træer. For denne kørsel har du angivet en kædelængde på 6.000.000.000 trin med prøveudtagning hvert 1.000 trin. Træfilen vil således indeholde 6.000 træer, og der skal derfor angives et burnin på 10 % i det øverste tekstfelt.
Den posteriore sandsynlighedsgrænseindstilling angiver en grænse, således at hvis en knude findes med mindre end denne frekvens i stikprøven af træer (dvs. har en posteriore sandsynlighed mindre end denne grænse), vil den ikke blive annoteret. Standardværdien på 0,5 betyder, at kun knuder, der ses i størstedelen af træerne, vil blive annoteret. Sæt denne til nul for at annotere alle knuder.
Måletræstypen angiver den trætopologi, der vil blive annoteret. Du kan enten vælge et bestemt træ fra en fil eller bede TreeAnnotator om at finde et træ i din prøve.Standardindstillingen, Maximum clade credibility tree, finder træet med det højeste produkt af den efterfølgende sandsynlighed for alle dets knuder.
For nodehøjder er standardindstillingen Common Ancestor Heights, som beregner højden af en node som gennemsnittet af MRCA-tiden for alle par af knuder i kladen. For træer med stor usikkerhed i topologien og dermed mange klader med lav støtte kan nogle andre metoder resultere i træer med negative grenlængder. I denne analyse er støtten for alle klader i det summariske træ meget høj, så det er ikke noget problem her.Vælg gennemsnitlige højder for knudehøjder. Dette sætter højderne (aldre) for hver enkelt knude i træet til den gennemsnitlige højde i hele stikprøven af træer for den pågældende klade.
Vælg den træfil, som BEAST har oprettet, som inputfil, og vælg en fil til output (her kaldte vi den Primates.MCC.tree). Tryk nu på Kør og vent på, at programmet er færdigt.
7 Visualisering af træestimatet
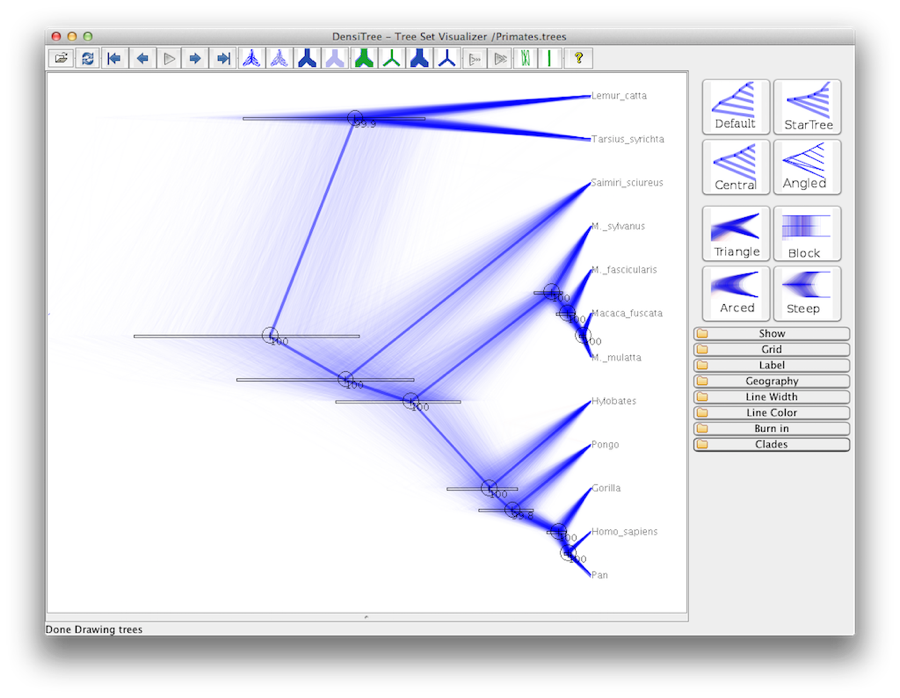
Sluttelig kan vi visualisere træet i et andet program kaldet FigTree. Kør dette program, og åbn filen Primates.MCC.tree ved hjælp af kommandoen Open (Åbn) i menuen File (Fil). Træet bør vises.Du kan nu prøve at vælge nogle af mulighederne i kontrolpanelet til venstre. Først og fremmest skal du bruge Trees-indstillingen i panelet og afkrydse Order nodes og vælge Ordering by decreasing. Prøv at vælge Node Bars (knudebjælker) for at få nodealdersfejlbjælker. Slå også Branch Labels til, og vælg Posterior for at få det til at vise den efterfølgende sandsynlighed for hver enkelt knude. Hvis du bruger en ikke-strenge urmodel, kan du under Appearance også fortælle FigTree, at grenene skal farvelægges efter hastigheden, og du bør ende op med noget, der ligner Figur 19.
Figur 19: Et skærmbillede af FigTree og DensiTree.
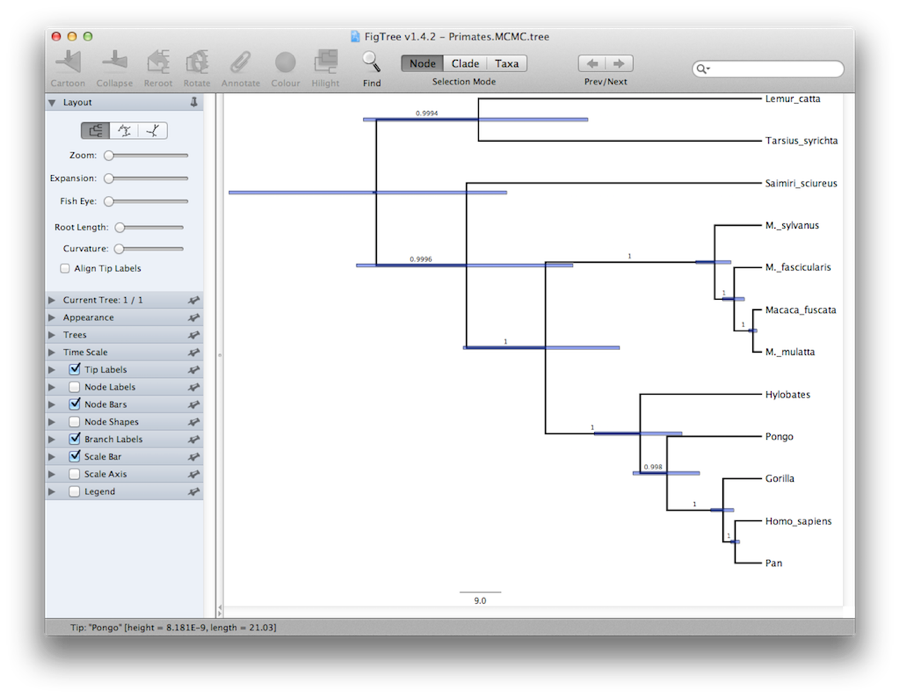
En alternativ visning af træet kan foretages med DensiTree, som er en del af Beast 2. Fordelen ved DensiTree er, at det er i stand til at visualisere både usikkerheden i knudehøjder og usikkerheden i topologien. for dette særlige datasæt er den dominerende topologi til stede i mere end 99 % af prøverne. Så vi konkluderer, at denne analyse resulterer i en meget høj konsensus om topologien (figur 19).
Spørgsmål
- Var udviklingshastigheden væsentligt forskellig blandt de forskellige slægtslinjer i træet?
- DensiTree har en kladebar (Menu Window/View clade toolbar) til at vise oplysninger om klader.
Hvad er støtten for kladen?
- Du kan gennemse topologierne i DensiTree ved hjælp af menuen Browse (Gennemse).Den mest populære topologi har en støtte på over 99 %.
Hvad er støtten for den næstmest populære topologi?
- Under hjælpemenuen viser DensiTree nogle oplysninger.
Hvor mange topologier er der i træsættet?
8 Sammenligning af dine resultater med prioriteten
Det er en god idé at genudføre analysen, mens du sampler fra prioriteten for at sikre dig, at interaktioner mellem prioriteter ikke påvirker dine prioritetsoplysninger. Interaktionen mellem priors kan være problematisk, især når der anvendes kalibreringer, da det betyder, at der sættes flere priors på træet.
Ved hjælp af BEAUti skal du opsætte den samme analyse, men under MCMC-indstillingerne skal du vælge indstillingen Sample from prior only (kun prøve fra prior). Dette vil give dig mulighed for at visualisere den fulde prioritetsfordeling i fravær af dine sekvensdata. Sammenfat træerne fra den fulde prioritetsfordeling, og sammenlign sammenfatningen med det efterfølgende sammenfatningstræ.
Divergenstidsestimering ved hjælp af “node-datering” af den type, der er beskrevet i dette kapitel, er blevet anvendt til at besvare en række forskellige spørgsmål inden for økologi og evolution. For eksempel blev node-datering med fossiler anvendt til at bestemme artsdiversiteten hos cycader, analysere udviklingshastigheden hos blomstrende planter og undersøge oprindelsen af cyanobakterier fra varme og kolde ørkener.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al.., Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond og Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), no. 1, 114. A Gelman, G Roberts og W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled og Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little og S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian og Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith og Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008), no. 5898, 86-89.
Dette dokument er oversat fra LATEX afHEVEA.