


Alecensa (Alectinib) ist ein Kinaseinhibitor zur Behandlung von Patienten mit anaplastischem Lymphomkinase (ALK)-positivem nicht-kleinzelligem Lungenkrebs (NSCLC).
Das Medikament wurde von dem zur Roche-Gruppe gehörenden Unternehmen Genentech in Zusammenarbeit mit Chugai Pharmaceutical entwickelt.
Alecensa wurde im Juli 2014 zunächst in Japan und im Dezember 2015 von der US-amerikanischen Food and Drug Administration (FDA) zugelassen.
Roche reichte im September 2015 bei der Europäischen Arzneimittelagentur (EMA) einen Zulassungsantrag für Alecensa ein.
Das Unternehmen erhielt im Februar 2017 von der Europäischen Kommission (EK) eine bedingte Zulassung für Alecensa als Monotherapie zur Behandlung von erwachsenen ALK-positiven Patienten mit fortgeschrittenem NSCLC.
Diese Patienten wurden zuvor mit Crizotinib, einem von Pfizer entwickelten NSCLC-Medikament, behandelt.
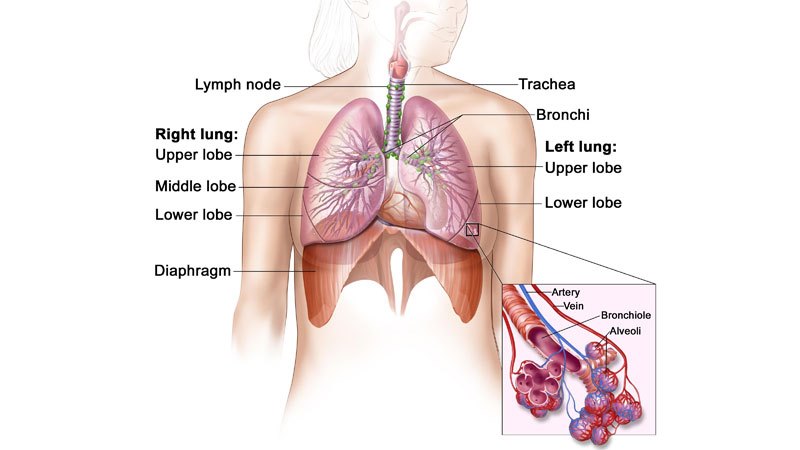
ALK-positiver fortgeschrittener NSCLC
Das nicht-kleinzellige Lungenkarzinom ist die häufigste Krebsart und macht mehr als 85 % der Lungenkrebserkrankungen aus. Weltweit sterben jährlich etwa 1,59 Millionen Menschen an NSCLC.
Die Krankheit tritt auf, wenn Zellen der Lunge abnormal werden und sich unkontrolliert zu entwickeln beginnen. Sie wird in der Regel in fortgeschrittenen Stadien diagnostiziert und ist dafür bekannt, dass sie in frühen Stadien schwer zu erkennen oder zu diagnostizieren ist.
Zu den Symptomen von Lungenkrebs gehören Husten, Kurzatmigkeit, Müdigkeit, Appetitlosigkeit und Gewichtsverlust.
ALK-positiver nicht-kleinzelliger Lungenkrebs tritt bei etwa 5 % der Patienten mit fortgeschrittenem nicht-kleinzelligem Lungenkrebs (NSCLC) auf, wobei weltweit schätzungsweise 75.000 Menschen pro Jahr die Diagnose erhalten.
Wirkungsmechanismus von Alecensa
Alecensa enthält einen Tyrosinkinase-Inhibitor, der die ALK-Phosphorylierung und die ALK-vermittelte Aktivierung der in NSCLC-Tumoren identifizierten nachgeschalteten Signalproteine verhindert.
Das Medikament ist derzeit in Form von 150mg-Kapseln zur oralen Verabreichung erhältlich.
Klinische Studien zu Alecensa
Die bedingte Marktzulassung von Alecensa durch die Europäische Kommission basierte auf zwei klinischen Studien der Phase I / II mit den Bezeichnungen NP28673 und NP28761.
Die klinische Studie NP28673 war eine globale, einarmige, offene, multizentrische Studie der Phase I / II, die die Sicherheit und Wirksamkeit von Alecensa bei 138 ALK-positiven NSCLC-Patienten untersuchte, deren Krankheit unter Crizotinib fortgeschritten war.
Die Ergebnisse zeigten, dass mit Alecensa behandelte Patienten eine Gesamtansprechrate (ORR) von 50.Die Ergebnisse zeigten, dass die Patienten, die mit Alecensa behandelt wurden, eine Gesamtansprechrate (ORR) von 50,8 % aufwiesen, die nach den RECIST-Kriterien (Response Evaluation Criteria In Solid Tumors) gemessen wurde.
Eine Bewertung durch die Prüfärzte zeigte, dass die Tumoren bei 51,4 % der Patienten, die Alecensa erhielten, zurückgingen.
Die Patienten sprachen im Median 15,2 Monate lang weiter an, während das mediane progressionsfreie Überleben (PFS) bei den Patienten, die Alecensa erhielten, 8,9 Monate betrug.
Die Ergebnisse zeigten auch, dass das Sicherheitsprofil von Alecensa dem in früheren Studien beobachteten ähnlich war.
Zu den unerwünschten Wirkungen, die bei ≥2% der mit dem Arzneimittel behandelten Patienten während der Studie gemeldet wurden, gehörten Dyspnoe, Anämie, Müdigkeit, erhöhter INR, Lungenembolie und Hyperbilirubinämie.
NP28761 war eine klinische Studie der Phase I/II, die in Nordamerika durchgeführt wurde. Es handelte sich um eine einarmige, offene, multizentrische Studie, die die Sicherheit und Wirksamkeit von Alecensa bei 87 ALK-positiven NSCLC-Patienten untersuchte, deren Krankheit auch während der Behandlung mit Crizotinib fortgeschritten war.
Die Ergebnisse der Studie zeigten, dass mit Alecensa behandelte Patienten bei einer Bewertung durch ein unabhängiges Prüfungskomitee, gemessen an den RECIST-Kriterien, eine ORR von 52,2 % aufwiesen.
Eine Bewertung durch die Prüfärzte zeigte, dass sich die Tumoren bei 52,9 % nach der Behandlung mit dem Arzneimittel zurückbildeten.
Die Probanden sprachen im Median 14,9 Monate lang auf die Behandlung an, und das mediane PFS lag bei den Patienten, die Alecensa erhielten, bei acht Monaten.
Die Ergebnisse zeigten, dass das Sicherheitsprofil von Alecensa dem in früheren Studien beobachteten ähnlich war.
Zu den häufigsten unerwünschten Ereignissen, die während der klinischen Studie gemeldet wurden, gehörten ein Anstieg der Muskelenzyme, erhöhte Leberenzyme, Kurzatmigkeit, erhöhte Triglyzeridwerte, niedrige Phosphat- und Kaliumwerte und eine verlängerte Zeit für eine partielle Blutverdickung.