Alexei Drummond, Andrew Rambaut, Remco Bouckaert und Walter Xie
1 Einleitung
Dieses Tutorial stellt die BEAST-Software für Bayes’sche Evolutionsanalysen anhand eines einfachen Tutorials vor. Das Tutorium beinhaltet die Co-Schätzung einer Gen-Phylogenie und der zugehörigen Divergenzzeiten in Gegenwart von Kalibrierungsinformationen aus fossilen Belegen.
Sie benötigen die folgende Software:
- BEAST – dieses Paket enthält das BEAST-Programm, BEAUti, TreeAnnotator und andere Hilfsprogramme. Diese Anleitung ist für BEAST v2.2.x geschrieben, das die Unterstützung für mehrere Partitionen bietet. Es kann heruntergeladen werden von http://www.beast2.org/.
- Tracer – dieses Programm wird verwendet, um die Ausgabe von BEAST (und anderen Bayesian MCMC-Programmen) zu untersuchen. Es fasst die Verteilungen der kontinuierlichen Parameter grafisch und quantitativ zusammen und liefert diagnostische Informationen. Zum Zeitpunkt der Erstellung dieses Dokuments ist die aktuelle Version v1.6. Sie kann von
http://tree.bio.ed.ac.uk/software/ heruntergeladen werden. - FigTree – dies ist eine Anwendung zur Anzeige und zum Ausdrucken von molekularen Phylogenien, insbesondere von solchen, die mitBEAST erstellt wurden. Zum Zeitpunkt der Erstellung dieses Artikels ist die aktuelle Version v1.4.2. Sie kann heruntergeladen werden von http://tree.bio.ed.ac.uk/software/.
Dieses Tutorial führt Sie durch die Analyse eines Alignments von Sequenzen aus zwölf Primatenarten (siehe Abbildung 1). Ziel ist es, die Phylogenie, die Evolutionsrate jeder Abstammungslinie und das Alter der unkalibrierten Stammesdivergenzen zu schätzen.
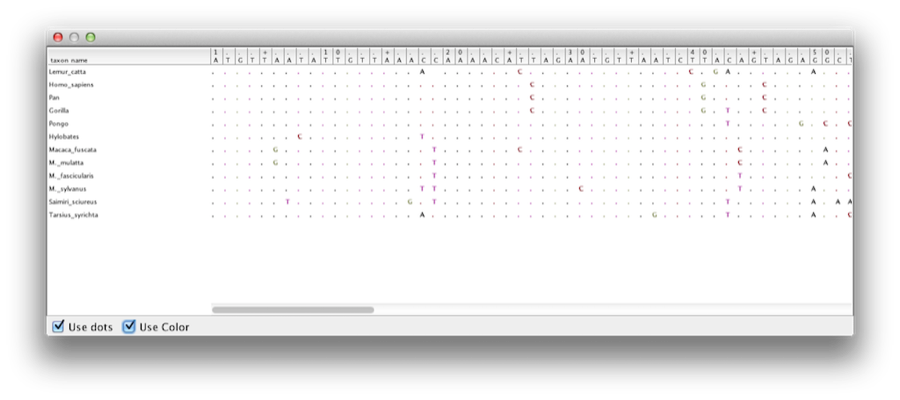
Abbildung 1: Teil des Alignments für Primaten.
Der erste Schritt besteht darin, eine NEXUS-Datei mit einem DATA- oder CHARACTERS-Block in eine BEAST-XML-Eingabedatei zu konvertieren. Dies geschieht mit dem Programm BEAUti (das für Bayesian Evolutionary Analysis Utility steht). Dabei handelt es sich um ein benutzerfreundliches Programm zur Einstellung des Evolutionsmodells und der Optionen für die MCMC-Analyse. Der zweite Schritt besteht darin, BEAST mit der von BEAUTi erzeugten Eingabedatei, die die Daten, das Modell und die Analyseeinstellungen enthält, auszuführen. Der letzte Schritt besteht darin, die Ausgabe von BEAST zu untersuchen, um Probleme zu diagnostizieren und die Ergebnisse zusammenzufassen.
2 BEAUti
Das Programm BEAUti ist ein benutzerfreundliches Programm zur Einstellung der Modellparameter für BEAST. Starten Sie BEAUti durch einen Doppelklick auf sein Symbol. Sobald BEAUti läuft, sieht es ähnlich aus, unabhängig davon, auf welchem Computersystem es ausgeführt wird. In diesem Tutorial wird die Mac OS X-Version verwendet, aber die Linux- und Windows-Versionen haben das gleiche Layout und die gleiche Funktionalität.
2.1 Laden der NEXUS-Datei
Um eine Ausrichtung im NEXUS-Format zu laden, wählen Sie einfach die Option Ausrichtung importieren… aus dem Menü Datei, oder ziehen Sie die Datei in die Mitte des Bereichs Partitionen.
Die Beispieldatei mit dem Namen primate-mtDNA.nex ist im Verzeichnis examples/nexus/ für Mac und Linux und examples/nexus/ für Windows in dem Verzeichnis verfügbar, in dem BEAST installiert wurde.
Ein Fenster zum Hinzufügen von Partitionen (Abbildung 2) wird angezeigt, wenn das entsprechende Paket installiert ist. Wenn Sie „reines“ BEAST 2 verwenden, können Sie mit dem nächsten Abschnitt fortfahren. Andernfalls wählen Sie Ausrichtung hinzufügen und klicken Sie auf OK, um fortzufahren.
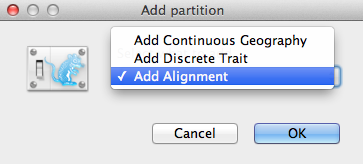
Abbildung 2: Fenster „Partition hinzufügen“ (erscheint nur, wenn zugehörige Pakete installiert sind).
Wenn es Überschneidungen bei der Codierung der Partitionen gibt, erscheint das Fenster mit der Warnmeldung (Abbildung 3). Lesen Sie es und klicken Sie auf OK, um fortzufahren.
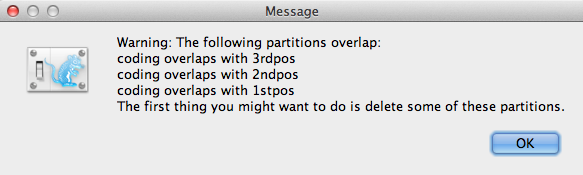
Abbildung 3: Warnmeldungsfenster (erscheint nur, wenn es Überschneidungen in den Partitionen gibt).
Nach dem Laden werden im Hauptfenster fünf Zeichenpartitionen angezeigt (Abbildung 4). Das Alignment ist in einen proteincodierenden Teil und einen nichtcodierenden Teil unterteilt, wobei der codierende Teil in die Codonpositionen 1, 2 und 3 unterteilt ist. Sie müssen die Partition „coding“ entfernen, bevor Sie mit dem nächsten Schritt fortfahren, da sie sich auf dieselben Nukleotide wie die Partitionen „1stpos“, „2ndpos“ und „3rdpos“ bezieht. Um die Partition ‚coding‘ zu entfernen, wählen Sie die Zeile aus und klicken Sie auf die Schaltfläche ‚-‚ am unteren Rand der Tabelle. Sie können die Ausrichtung anzeigen, indem Sie auf die Partition doppelklicken.
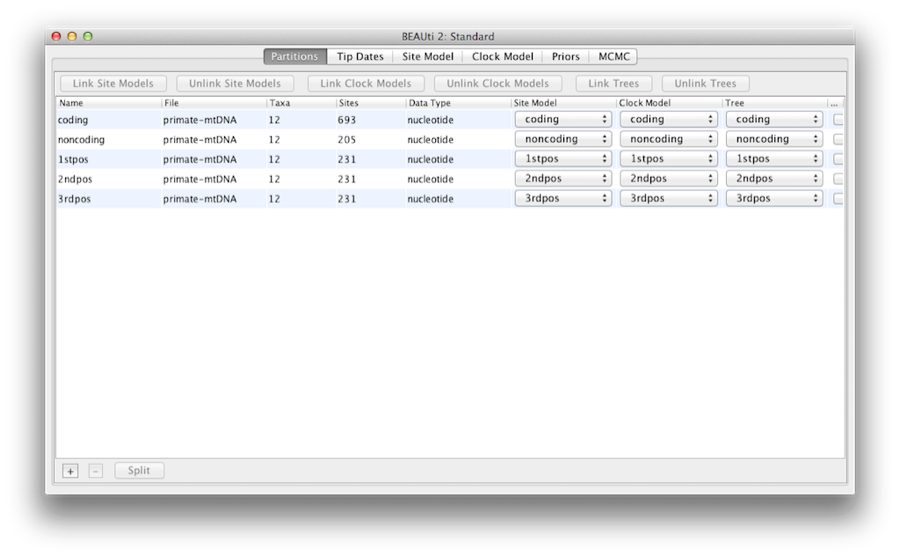
Abbildung 4: Ein Screenshot der Registerkarte „Daten“ in BEAUti. Dieser und alle folgenden Screenshots wurden auf einem Apple-Computer mit Mac OS X aufgenommen und sehen auf anderen Betriebssystemen etwas anders aus.
Verknüpfung/Entknüpfung von Partitionsmodellen
Abbildung 5: Ein Screenshot der Registerkarte Partitionen in BEAUti nach der Verknüpfung und Umbenennung des Uhrenmodells und des Baums.
Da die Sequenzen miteinander verknüpft sind (d. h. sie stammen alle aus dem mitochondrialen Genom, von dem angenommen wird, dass es bei Vögeln und Säugetieren nicht rekombiniert wird), haben sie dieselbe Abstammung, so dass die Partitionen im Modell denselben Zeitbaum haben sollten. Der Einfachheit halber gehen wir auch davon aus, dass die Partitionen für jeden Zweig dieselbe Evolutionsrate und damit dasselbe „Uhrenmodell“ haben. Wir beschränken unsere Modellierung der Ratenheterogenität auf die Heterogenität zwischen den Standorten innerhalb jeder Partition und lassen auch zu, dass die Partitionen unterschiedliche mittlere Evolutionsraten haben.
Also müssen wir an dieser Stelle das Uhrenmodell und den Baum miteinander verbinden. Wählen Sie im Bereich Partitionen alle vier Partitionen in der Tabelle aus (oder keine, standardmäßig sind alle Partitionen betroffen) und klicken Sie auf die Schaltfläche Bäume verknüpfen und dann auf die Schaltfläche Uhrenmodelle verknüpfen (siehe Abbildung 5). Klicken Sie dann auf das erste Dropdown-Menü in der Spalte Taktmodell und benennen Sie das gemeinsame Taktmodell in „Uhr“ um. Ebenso benennen Sie den gemeinsamen Baum in „Baum“ um. Dadurch werden die folgenden Optionen und die erzeugten Protokolldateien leichter zu lesen sein.
2.2 Einstellen des Substitutionsmodells
Der nächste Schritt ist die Einrichtung des Substitutionsmodells. Wählen Sie die Registerkarte „Site Models“ im oberen Teil des Hauptfensters (wir überspringen die Registerkarte „Tip Dates“, da alle Taxa aus zeitgenössischen Proben stammen). Daraufhin werden die Einstellungen des Evolutionsmodells für BEAST angezeigt. Die verfügbaren Optionen hängen davon ab, ob es sich bei den Daten um Nukleotide oder Aminosäuren, binäre Daten oder allgemeine Daten handelt. Die Einstellungen, die nach dem Laden des Primaten-Nukleotid-Alignments angezeigt werden, sind die Standardwerte für Nukleotiddaten, so dass wir einige Änderungen vornehmen müssen.
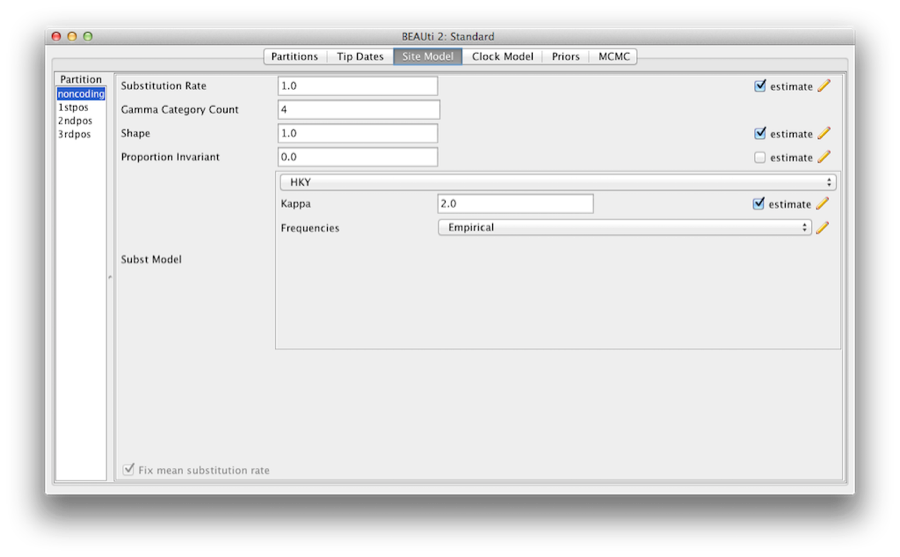
Abbildung 6: Ein Screenshot der Registerkarte „Site Model“ in BEAUti.
Die meisten der Modelle sollten Ihnen bekannt sein. Setzen Sie zunächst die Gamma-Kategorie Count auf 4 und kreuzen Sie dann das Feld „Schätzung“ für den Parameter Shape an. Dies ermöglicht die Modellierung der Ratenvariation zwischen den Standorten in jeder Partition. Beachten Sie, dass 4 bis 6 Kategorien für die meisten Datensätze ausreichend sind, während mehr Kategorien mehr Zeit für die Berechnung benötigen, aber nur wenig zusätzlichen Nutzen bringen. Wir lassen den Eintrag Proportion Invariant auf Null gesetzt.
Wählen Sie dann HKY aus dem Dropdown-Menü Subst Model. Idealerweise sollte ein Substitutionsmodell gewählt werden, das für jede Partition am besten zu den Daten passt, aber der Einfachheit halber verwenden wir hier HKY für alle Partitionen. Wählen Sie außerdem Empirisch aus dem Dropdown-Menü Häufigkeiten. Dadurch werden die Häufigkeiten auf die in den Daten beobachteten Anteile festgelegt (für jede Partition einzeln, sobald wir die Verknüpfung der Standortmodelle aufheben). Dieser Ansatz bedeutet, dass wir eine gute Anpassung an die Daten erhalten können, ohne diese Parameter explizit zu schätzen. Wir tun dies hier nur, um die Protokolldateien etwas kürzer und in späteren Teilen der Übung besser lesbar zu machen.
Abbildung 7: Klonkonfiguration von einem Standortmodell zu anderen.
Schließlich markieren Sie das Kästchen „Schätzung“ für den Parameter „Substitutionsrate“ und aktivieren Sie das Kästchen „Mittlere Mutationsrate fixieren“. Dadurch werden die relativen Raten der einzelnen Partitionen für die unverknüpften Site-Modelle geschätzt (Abbildung 6).
Halten Sie schließlich die Umschalttaste gedrückt, um alle Site-Modelle auf der linken Seite auszuwählen, und klicken Sie auf OK, um die Einstellung von „noncoding“ in „1stpos“, „2ndpos“ und „3rdpos“ zu klonen (Abbildung 7). Gehen Sie die einzelnen Standortmodelle durch, wie Sie sehen können, sind ihre Konfigurationen jetzt gleich.
2.3 Einstellung des Uhrenmodells
Der nächste Schritt ist die Auswahl der Registerkarte Uhrenmodelle oben im Hauptfenster. Hier wählen wir das molekulare Uhrenmodell aus. Für diese Übung belassen wir die Auswahl auf dem Standardwert einer strengen molekularen Uhr, da diese Daten sehr taktähnlich sind und keine Ratenvariation zwischen den Zweigen in das Modell aufgenommen werden muss.
Um die Taktähnlichkeit zu testen, kann man (i) die Analyse mit einem entspannten Taktmodell durchführen und prüfen, wie viel Variation zwischen den Raten durch die Daten impliziert wird (siehe Variationskoeffizient für mehr dazu), oder(ii) einen Modellvergleich zwischen einem strikten und entspannten Takt unter Verwendung von Pathsampling durchführen, oder (iii) ein zufälliges lokales Taktmodell verwenden, das explizit berücksichtigt, ob jeder Zweig im Baum seine eigene Zweigrate benötigt.
2.4 Prioritäten
Die Registerkarte „Prioritäten“ ermöglicht die Festlegung von Prioritäten für jeden Parameter im Modell. Die in den Registerkarten „Standortmodell“ und „Uhrenmodell“ getroffenen Modellauswahlen führen zur Einbeziehung verschiedener Parameter in das Modell, die auf der Registerkarte „Prioritäten“ angezeigt werden (siehe Abbildung 8).
Abbildung 8: Screenshot der Registerkarte „Prioritäten“ in BEAUti.
Hier geben wir auch an, dass wir das kalibrierte Yule-Modell als Baumprior verwenden möchten. Das Yule-Modell ist ein einfaches Modell der Speziation, das im Allgemeinen besser geeignet ist, wenn Sequenzen von verschiedenen Arten betrachtet werden. Wählen Sie das kalibrierte Yule-Modell aus dem Dropdown-Menü Baumprior aus.
2.4.1 Definieren des Kalibrierungsknotens
Wir müssen nun eine Prioritätsverteilung für den kalibrierten Knoten angeben, die auf unserem Fossilwissen basiert. Dies wird als Kalibrierung unseres Baumes bezeichnet. Um eine zusätzliche Priorität zu definieren, klicken Sie auf die kleine Schaltfläche + unter der Liste der Prioritäten. Wenn sie in Ihrer Ansicht nicht sichtbar ist, scrollen Sie bitte im Panel nach unten, um die Schaltfläche + zu finden. Daraufhin wird ein Dialogfeld angezeigt, in dem Sie eine Teilmenge der Taxa im phylogenetischen Baum definieren können. Sobald Sie eine Taxa-Gruppe erstellt haben, können Sie später Kalibrierungsinformationen für ihren jüngsten gemeinsamen Vorfahren (MRCA) hinzufügen.
Benennen Sie den Taxa-Satz, indem Sie den Eintrag für die Bezeichnung des Taxa-Satzes ausfüllen. Nennen Sie es Mensch-Schimpanse, da es die Taxa für Homo sapiens und Pan enthalten wird. In der Liste unten sehen Sie die verfügbaren Taxa. Wählen Sie nacheinander die beiden Taxa aus und drücken Sie die Pfeiltaste > >. (Da es sich um einen kalibrierten Knoten handelt, der in Verbindung mit dem kalibrierten Yule-Prior verwendet werden soll, muss die Monophylie erzwungen werden, aktivieren Sie also das Kontrollkästchen Monophyletisch. Dadurch wird die Baumtopologie so eingeschränkt, dass die Gruppierung Mensch-Schimpanse im Verlauf der MCMC-Analyse monophyletisch bleibt.
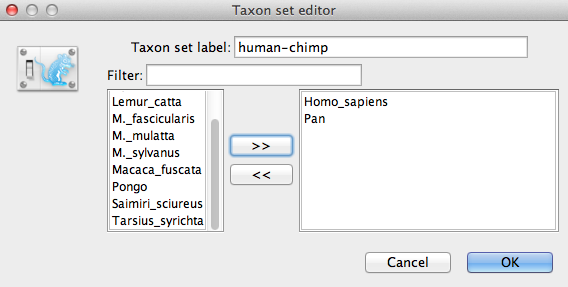
Abbildung 9: Taxon-Set-Editor in BEAUti.
Um die Kalibrierungsinformationen zu kodieren, müssen wir eine Verteilung für die MRCA von Mensch-Schimpanse angeben.Wählen Sie die Log-Normal-Verteilung aus dem Dropdown-Menü rechts neben der neu hinzugefügten Mensch-Schimpanse.prior. Klicken Sie auf das schwarze Dreieck, und es erscheint ein Diagramm der Wahrscheinlichkeitsdichtefunktion zusammen mit den Parametern für die Log-Normal-Verteilung. Wir legen M=1,78 und S=0,085 fest, was eine Verteilung mit einem Zentrum bei etwa 6 Millionen Jahren und einer Standardabweichung von etwa 0,5 Millionen Jahren ergibt. Damit ergibt sich ein zentraler Wahrscheinlichkeitsbereich von 95 %, der 5-7 Mya abdeckt. Dies entspricht in etwa der aktuellen Konsensschätzung des Datums des jüngsten gemeinsamen Vorfahren von Menschen und Schimpansen (Abbildung 10).
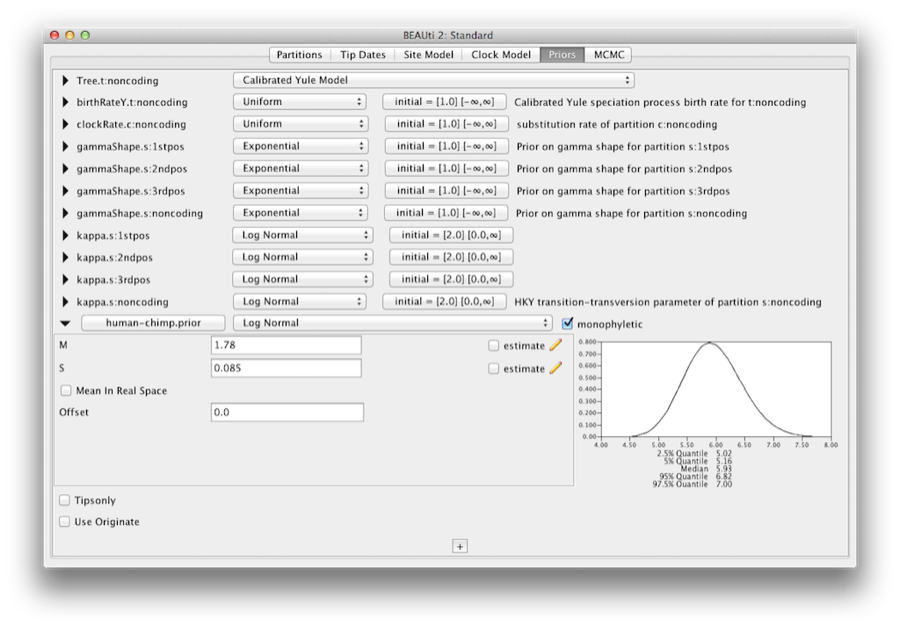
Abbildung 10: Bildschirmfoto der Kalibrierungsprioroptionen im Priors-Panel in BEAUti.
Wir sollten uns davon überzeugen, dass die im Prioren-Panel angezeigten Prioren wirklich die Informationen widerspiegeln, die wir über die Parameter des Modells haben. Schließlich werden wir auch einige diffuse „uninformative“, aber angemessene Prioritäten für die Gesamtrate der molekularen Uhr (clockRate) und die Speziationsrate (birthRateY) des Yule-Baums angeben. Wählen Sie für jeden dieser Prioren Gamma aus dem Dropdown-Menü und erweitern Sie die Ansicht mit Hilfe der Pfeiltaste, um die Parameter des Gamma-Priors anzuzeigen. Setzen Sie sowohl für die Clock-Rate als auch für die Yule-Geburtsrate den Alpha(shape)-Parameter auf 0,001 und den Beta(scale)-Parameter auf 1000 (Abbildung 11).
Standardmäßig hat jeder der Gamma-Shape-Parameter eine exponentielle Priordistribution mit einem Mittelwert von 1. Das bedeutet (siehe Abbildung 3.7), dass wir eine gewisse Variation erwarten. Die Kappa-Parameter für das HKY-Modell haben standardmäßig eine Lognormal(1,1.25)-Priorverteilung, die weitgehend mit den empirischen Erkenntnissen über den Bereich realistischer Werte für Übergangs-/Transversionsbias übereinstimmt. Diese Standardprioritäten werden beibehalten, da sie für diese spezielle Analyse geeignet sind.
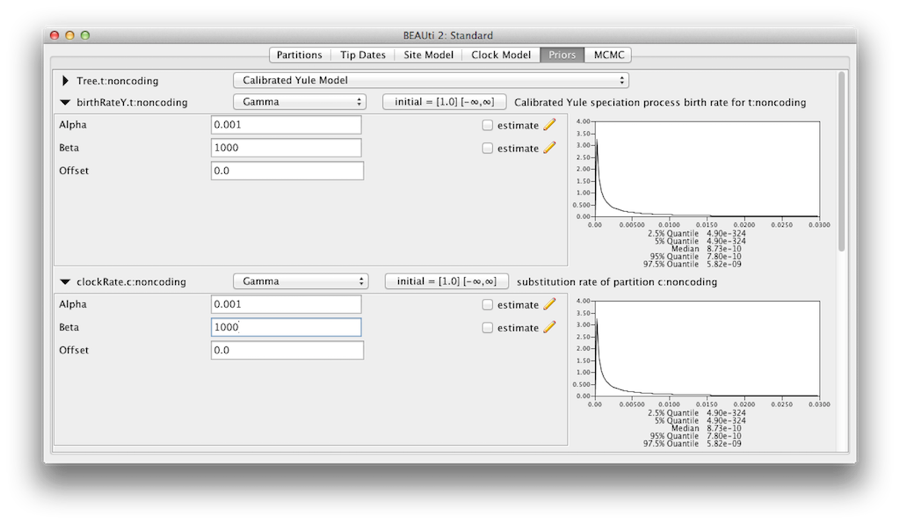
Abbildung 11: Gamma-Prior.
2.5 Einstellen der MCMC-Optionen
Die nächste Registerkarte, MCMC, bietet allgemeinere Einstellungen zur Steuerung der Länge des MCMC-Laufs und der Dateinamen.
Zunächst haben wir die Kettenlänge. Dies ist die Anzahl der Schritte, die die MCMC in der Kette macht, bevor sie beendet wird. Wie lang diese sein sollte, hängt von der Größe des Datensatzes, der Komplexität des Modells und der Qualität der gewünschten Antwort ab. Der Standardwert von 10.000.000 ist völlig willkürlich und sollte entsprechend der Größe Ihres Datensatzes angepasst werden. Für diesen Datensatz setzen wir die Kettenlänge auf 6.000.000, da dies auf den meisten modernen Computern relativ schnell geht (ein paar Minuten).
Das Feld Store Every legt fest, wie oft der Zustand in einer Datei gespeichert wird. Die periodische Speicherung des Zustands ist nützlich, wenn die Computerumgebung nicht sehr zuverlässig ist und ein BEAST-Lauf unterbrochen werden kann. Mit einer gespeicherten Kopie des aktuellen Zustands können Sie die Kette fortsetzen, anstatt wieder von vorne zu beginnen, so dass Sie nicht noch einmal durch das Einbrennen gehen müssen.Das Feld Pre Burnin gibt die Anzahl der Proben an, die nicht gleich zu Beginn der Analyse protokolliert werden. Wir belassen die Felder Store Every und Pre Burnin auf ihren Standardwerten. Darunter befinden sich die Details der Protokolldateien. Jede dieser Dateien kann durch Anklicken des schwarzen Dreiecks erweitert werden.
Die nächsten Optionen geben an, wie oft die Parameterwerte in der Markovchain auf dem Bildschirm angezeigt und in der Protokolldatei aufgezeichnet werden sollen.Die Bildschirmausgabe dient lediglich der Überwachung des Programmfortschritts und kann auf einen beliebigen Wert eingestellt werden (wenn sie zu klein eingestellt ist, wird das Programm durch die schiere Menge der auf dem Bildschirm angezeigten Informationen verlangsamt). Für die Protokolldatei sollte der Wert in Relation zur Gesamtlänge der Kette gesetzt werden. Eine zu häufige Abtastung führt zu sehr großen Dateien mit wenig zusätzlichem Nutzen für die Genauigkeit der Analyse. Bei zu seltenen Stichproben werden in der Protokolldatei nicht genügend Informationen über die Verteilungen der Parameter aufgezeichnet. Wahrscheinlich möchten Sie nicht mehr als 10.000 Stichproben speichern, so dass der Wert nicht kleiner als Kettenlänge / 10.000 sein sollte.
Für diese Übung werden wir die Häufigkeit des Trace-Logs und des Tree-Logs auf 1.000 und die des Screen-Logs auf 10.000 setzen und Primates.log als Dateinamen für die Trace-Log-Datei und Primates.trees als Dateinamen für die Tree-Log-Datei angeben.Vergewissern Sie sich, dass das Dateinamensfeld des Bildschirmprotokolls leer bleibt, sonst wird das Bildschirmprotokoll nicht auf den Bildschirm geschrieben.
- Wenn Sie das Windows-Betriebssystem verwenden, empfehlen wir Ihnen, an beide Dateien (also Primates.log.txt und Primates.trees.txt) die Endung .txt anzuhängen, damit Windows sie als Textdateien erkennt.
2.6 Erzeugen der BEAST-XML-Datei
Wir sind nun bereit, die BEAST-XML-Datei zu erstellen. Wählen Sie dazu im Menü „Datei“ die Option „Speichern“. Überprüfen Sie die Standardprioritäten und speichern Sie die Datei unter einem geeigneten Namen (normalerweise endet der Dateiname mit .xml, z. B. Primates.xml). Jetzt können Sie die Datei in BEAST ausführen.
3 BEAST ausführen
Abbildung 12: Ein Screenshot von BEAST.
Nun starten Sie BEAST, und wenn es nach einer Eingabedatei fragt, geben Sie Ihre neu erstellte XML-Datei als Eingabe ein. BEAST wird dann so lange laufen, bis es die Informationen auf dem Bildschirm ausgegeben hat. Die eigentlichen Ergebnisdateien werden auf der Festplatte an demselben Ort wie Ihre Eingabedatei gespeichert. Die Ausgabe auf dem Bildschirm sieht dann etwa so aus:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Beachten Sie, dass es am Anfang einige nützliche Informationen über die Ausrichtungen und die verwendeten Baumwahrscheinlichkeiten gibt. Außerdem werden zu Beginn des Durchlaufs alle für die Analyse relevanten Zitate genannt, die leicht in Manuskripte kopiert werden können, die über die Analyse berichten. Am Ende wird eine Operatoranalyse gedruckt, die alle in der Analyse verwendeten Operatoren auflistet und angibt, wie oft der Operator ausprobiert, akzeptiert und abgelehnt wurde (siehe Spalten #total, #accept bzw. #reject). Die Akzeptanzrate ist der Anteil der Fälle, in denen ein Operator akzeptiert wird, wenn er für einen Vorschlag ausgewählt wird. Im Allgemeinen zeigt eine hohe Akzeptanzrate, z. B. über 0,5, dass die Vorschläge konservativ sind und den Parameterraum nicht effizient erkunden. Andererseits deutet eine niedrige Akzeptanzrate darauf hin, dass die Vorschläge zu aggressiv sind und fast immer zu einem Zustand führen, der aufgrund seines niedrigen Posteriorwerts abgelehnt wird.Sowohl zu hohe als auch zu niedrige Akzeptanzraten führen zu niedrigen ESS-Werten. Für viele (aber nicht alle) in BEAST implementierten Operatoren wird eine Akzeptanzrate von 0,234 angestrebt (auf der Grundlage sehr begrenzter Nachweise).
Einige Operatoren haben einen Tuning-Parameter, zum Beispiel den Skalierungsfaktor des ascale-Parameters. Wenn die endgültige Akzeptanzrate nicht in der Nähe des Ziels liegt, schlägt BEAST einen neuen Wert für den Tuning-Parameter vor, der in der Operatoranalyse ausgegeben wird. In diesem Fall sind alle Akzeptanzraten für die Operatoren mit Tuning-Parametern gut. Zu den Operatoren ohne Abstimmparameter gehören in dieser Analyse die Wide-Exchange- und Wilson-Balding-Operatoren. Diese beiden Operatoren versuchen, die Topologie des Baumes mit großen Schritten zu verändern, aber da die Daten überwiegend eine einzige Topologie unterstützen, werden diese radikalen Vorschläge fast immer abgelehnt.
4 Analyse der Ergebnisse
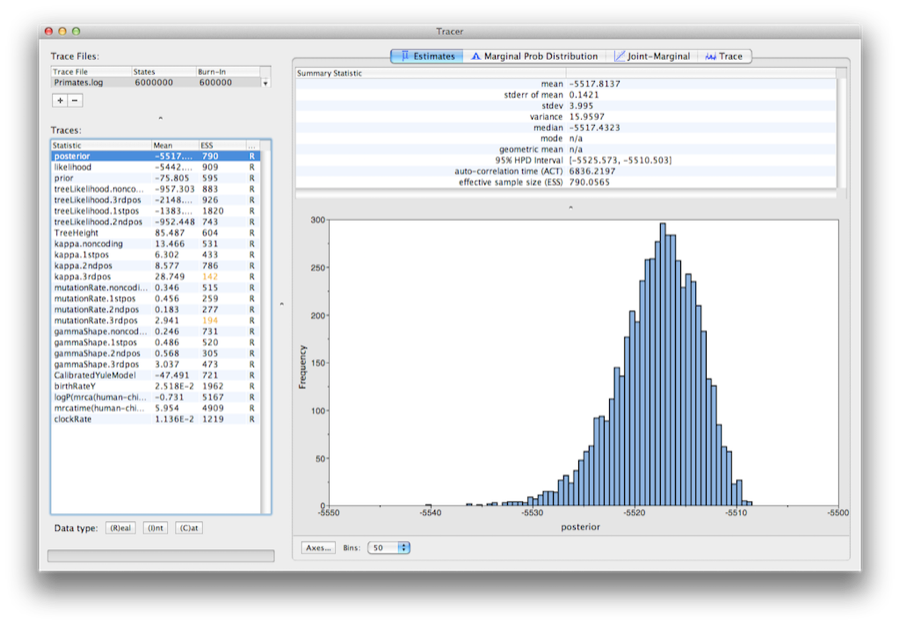
Abbildung 13: Ein Bildschirmfoto von Tracer v1.6.
Starten Sie das Programm Tracer, um die Ausgabe von BEAST zu analysieren. Wenn sich das Hauptfenster geöffnet hat, wählen Sie Import Trace File… aus dem Menü File und wählen Sie die von BEAST erstellte Datei Primates.log (Abbildung 13).
Denken Sie daran, dass MCMC ein stochastischer Algorithmus ist, so dass die tatsächlichen Zahlen nicht genau mit den in der Abbildung dargestellten übereinstimmen werden.
Auf der linken Seite befindet sich eine Liste der verschiedenen Größen, die BEAST in einer Datei aufgezeichnet hat. Es gibt Spuren für das Posterior (dies ist der natürliche Logarithmus des Produkts aus der Baumwahrscheinlichkeit und der Prior-Dichte) und die kontinuierlichen Parameter. Wenn Sie auf der linken Seite eine Spur auswählen, werden auf der rechten Seite je nach gewählter Registerkarte Analysen für diese Spur angezeigt. Beim ersten Öffnen wird die „posteriore“ Spur ausgewählt und verschiedene Statistiken dieser Spur werden unter der Registerkarte „Schätzungen“ angezeigt. rechts oben im Fenster befindet sich eine Tabelle mit berechneten Statistiken für die ausgewählte Spur.
Wählen Sie den Parameter clockRate in der linken Liste, um die durchschnittliche Evolutionsrate zu betrachten (gemittelt über den gesamten Baum und alle Standorte). Tracer stellt ein (marginales posteriores) Histogramm für die ausgewählte Statistik dar und gibt auch zusammenfassende Statistiken wie Mittelwert und Median an. Das 95% HPD steht für das Intervall mit der höchsten Posterior-Dichte und stellt das kompakteste Intervall für den ausgewählten Parameter dar, das 95% der Posterior-Wahrscheinlichkeit enthält. Es kann als Bayessches Analogon zu einem Konfidenzintervall betrachtet werden. Der TreeHeight-Parameter gibt die marginale Posterior-Verteilung des Alters der Wurzel des gesamten Baumes an.
Wählen Sie den TreeHeight-Parameter aus und klicken Sie dann bei gedrückter Strg-Taste auf mrcatime(human-chimp) (Befehlsklick unter Mac OS X). Daraufhin werden das Alter der Wurzel und die Kalibrierungs-MRCA, die wir zuvor in BEAUti angegeben haben, angezeigt. Sie können sich vergewissern, dass die Divergenz, die wir zur Kalibrierung des Baums (mrcatime(human-chimp)) verwendet haben, eine Posterior-Verteilung hat, die mit der von uns angegebenen Prior-Verteilung übereinstimmt (Abbildung 14).
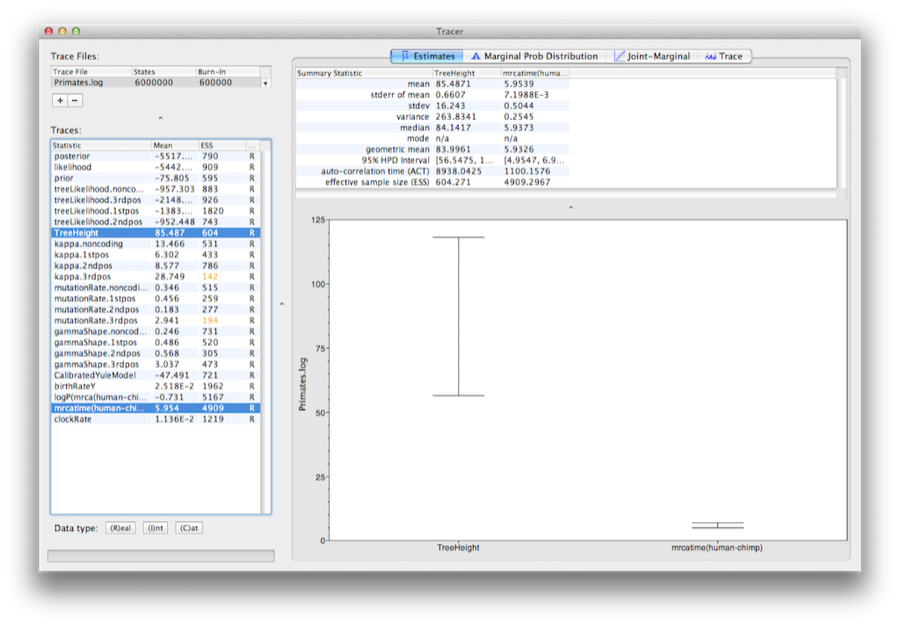
Abbildung 14: Ein Screenshot der 95%-HPD-Intervalle der Wurzelhöhe und der vom Benutzer angegebenen (human-chimp) MRCA in Tracer.
5 Marginale posteriore Schätzungen
Um die relativen Raten für die vier Partitionen anzuzeigen, wählen Sie den Parameter mutationRate für jede der vier Partitionen und wählen Sie die Registerkarte marginale Dichte in Tracer.Abbildung 15 zeigt die marginalen Dichten für die relativen Substitutionsraten. Die Darstellung zeigt, dass die Codonpositionen 1 und 2 deutlich unterschiedliche Raten aufweisen (0,456 versus 0,183) und beide weit langsamer sind als Codonposition 3 mit einer relativen Rate von 2,941. Die nichtcodierende Partition hat eine Rate, die zwischen den Codonpositionen 1 und 2 liegt (0,346). Zusammengenommen deutet dieses Ergebnis auf eine starke reinigende Selektion sowohl in den kodierenden als auch in den nichtkodierenden Regionen des Alignments hin.
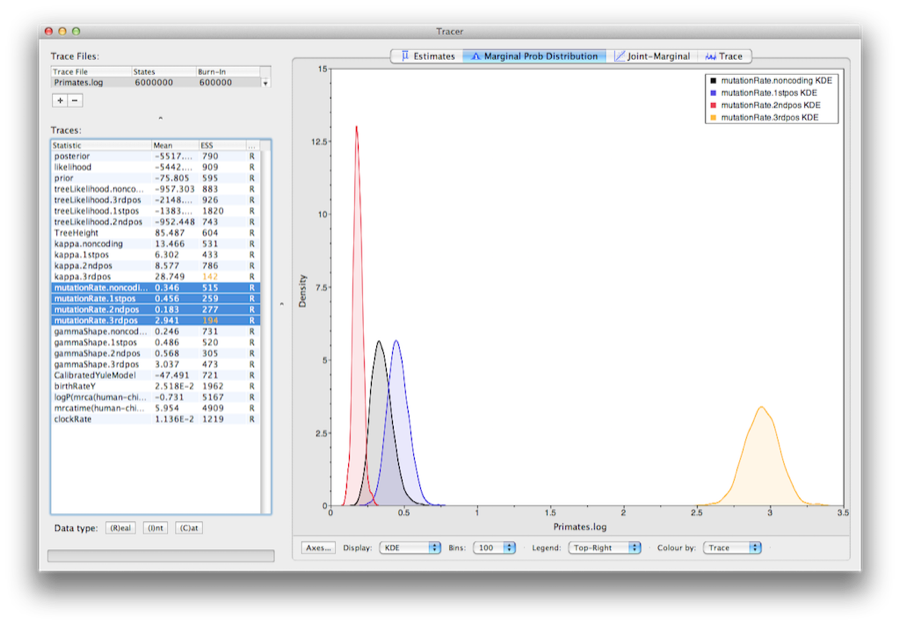
Abbildung 15: Ein Screenshot der marginalen Posterior-Dichten der relativen Substitutionsraten der vier Partitionen (relativ zur standortgewichteten mittleren Rate).
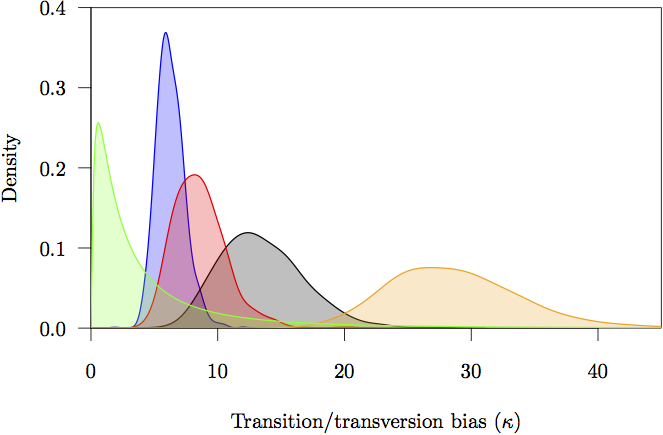
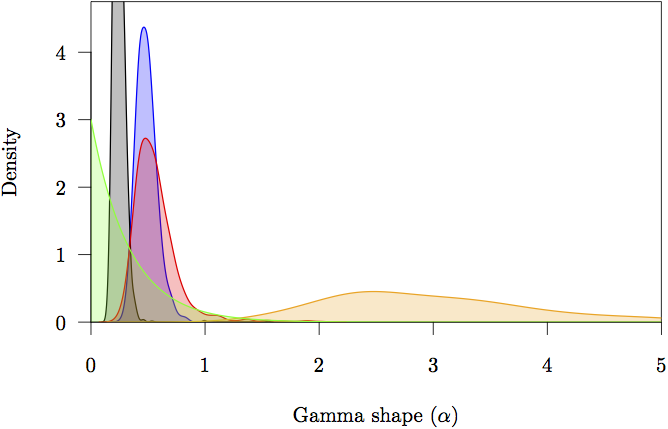
Abbildung 16: Die marginalen Prior- und Posterior-Dichten für die Form (α) Parameter. Der Prior ist in grau dargestellt. Die Posterior-Dichte-Schätzung für jede Partition ist ebenfalls dargestellt: nicht kodierende (orange) und erste (rot), zweite (grün) und dritte (blau) Codon-Positionen.
Abbildung 17: Die marginalen Prior- und Posterior-Dichten für die Übergangs/Transversions-Bias (κ)-Parameter. Der Prior ist in grau dargestellt. Die Posterior-Dichte-Schätzung für jede Partition ist ebenfalls dargestellt: nicht kodierende (orange) und erste (rot), zweite (grün) und dritte (blau) Codon-Positionen.
Fragen
Wie hoch ist die geschätzte Rate der molekularen Evolution für diesen Genbaum (einschließlich des 95% HPD-Intervalls)?
Welche Fehlerquellen sind in dieser Schätzung enthalten?
Wie alt ist die Wurzel des Baumes (geben Sie den Mittelwert und den 95%-HPD-Bereich an)?
6 Erhalt einer Schätzung des phylogenetischen Baumes
BEAST erzeugt auch eine posteriore Stichprobe phylogenetischer Zeitbäume zusammen mit seiner Stichprobe von Parameterschätzungen. Diese müssen mit Hilfe des Programms TreeAnnotator zusammengefasst werden. Dieses Programm sucht aus der Menge der Bäume denjenigen heraus, der am besten unterstützt wird. Dieser repräsentative zusammenfassende Baum wird dann mit dem mittleren Alter aller Knoten und den entsprechenden 95%-HPD-Bereichen kommentiert. Es berechnet auch die posteriore Kladenwahrscheinlichkeit für jeden Knoten. Starten Sie das Programm TreeAnnotator und richten Sie es wie in Abbildung 18 dargestellt ein.
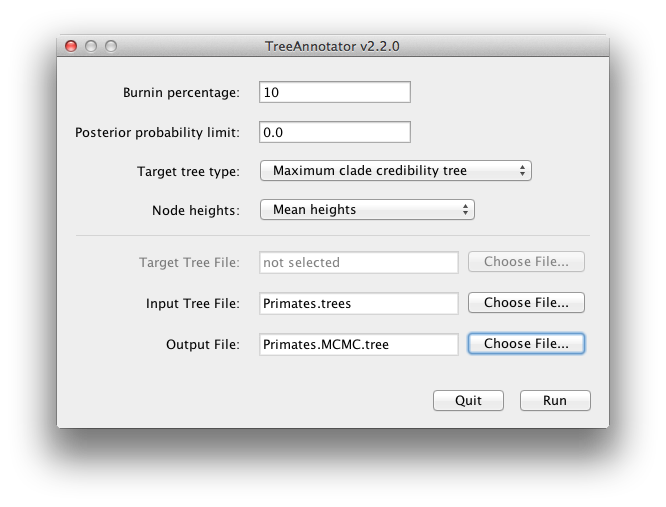
Abbildung 18: Ein Screenshot von TreeAnnotator.
Das Burnin ist die Anzahl der Bäume, die vom Anfang der Stichprobe entfernt werden sollen. Im Gegensatz zu Tracer, das die Anzahl der Schritte als Burnin angibt, müssen Sie in TreeAnnotator die tatsächliche Anzahl der Bäume angeben. Für diesen Lauf haben Sie eine Kettenlänge von 6.000.000 Schritten angegeben, wobei alle 1.000 Schritte gesampelt werden. Daher wird die Baumdatei 6.000 Bäume enthalten, so dass im oberen Textfeld ein 10%iger Burnin angegeben werden muss.
Die Option „Posterior probability limit“ legt einen Grenzwert fest, der besagt, dass ein Knoten, der in der Baumstichprobe weniger häufig gefunden wird (d.h. eine Posteriorwahrscheinlichkeit hat, die unter diesem Grenzwert liegt), nicht annotiert wird. Der Standardwert von 0,5 bedeutet, dass nur Knoten, die in der Mehrheit der Bäume vorkommen, annotiert werden. Setzen Sie diesen Wert auf Null, um alle Knoten zu annotieren.
Der Zielbaumtyp gibt die Baumtopologie an, die annotiert werden soll. Sie können entweder einen bestimmten Baum aus einer Datei auswählen oder TreeAnnotator auffordern, einen Baum in Ihrer Stichprobe zu finden.
Die Standardoption Maximale Kladenglaubwürdigkeitsbaum findet den Baum mit dem höchsten Produkt der Posteriorwahrscheinlichkeit aller seiner Knoten.
Für die Knotenhöhen ist die Standardeinstellung Common Ancestor Heights, die die Höhe eines Knotens als Mittelwert der MRCA-Zeit aller Knotenpaare innerhalb der Klade berechnet. Bei Bäumen mit großer Unsicherheit in der Topologie und somit vielen Kladen mit geringer Unterstützung können einige andere Methoden zu Bäumen mit negativen Zweiglängen führen. In dieser Analyse ist die Unterstützung für alle Kladen im zusammenfassenden Baum sehr hoch, so dass dies hier kein Problem darstellt.Wählen Sie mittlere Höhen für Knotenhöhen. Dies setzt die Höhen (Alter) jedes Knotens im Baum auf die mittlere Höhe der gesamten Stichprobe von Bäumen für diese Klade.
Wählen Sie für die Eingabedatei die von BEAST erstellte Baumdatei und wählen Sie eine Datei für die Ausgabe (hier haben wir sie Primates.MCC.tree genannt). Drücken Sie nun Ausführen und warten Sie, bis das Programm beendet ist.
7 Visualisierung der Baumschätzung
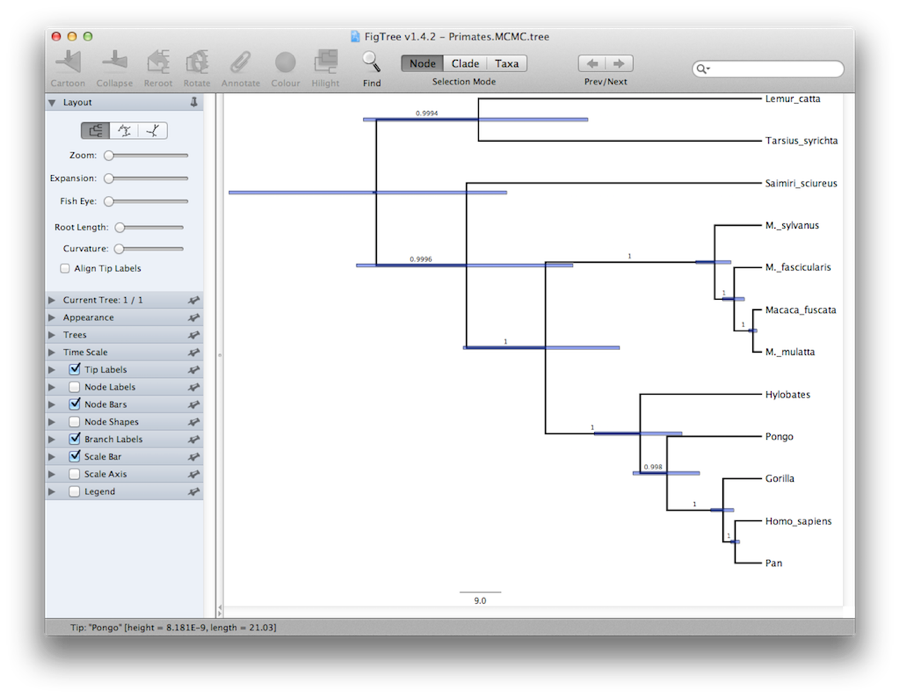
Schließlich können wir den Baum in einem anderen Programm namens FigTree visualisieren. Starten Sie dieses Programm und öffnen Sie die Datei Primates.MCC.tree mit dem Befehl Öffnen im Menü Datei. Der Baum sollte erscheinen. Sie können nun versuchen, einige der Optionen im Bedienfeld auf der linken Seite auszuwählen. Wählen Sie zunächst die Option Bäume im Bedienfeld aus, markieren Sie die Option Knoten ordnen und wählen Sie die Option Ordnen nach abnehmendem Wert. Wählen Sie Knotenbalken, um Fehlerbalken für das Alter der Knoten zu erhalten. Aktivieren Sie außerdem die Option Zweigbeschriftungen und wählen Sie Posterior, um die Posterior-Wahrscheinlichkeit für jeden Knoten anzuzeigen. Wenn Sie ein nicht striktes Uhrmodell verwenden, können Sie FigTree unter Erscheinungsbild auch anweisen, die Zweige nach der Rate zu färben, so dass Sie am Ende ein ähnliches Ergebnis wie in Abbildung 19 erhalten.
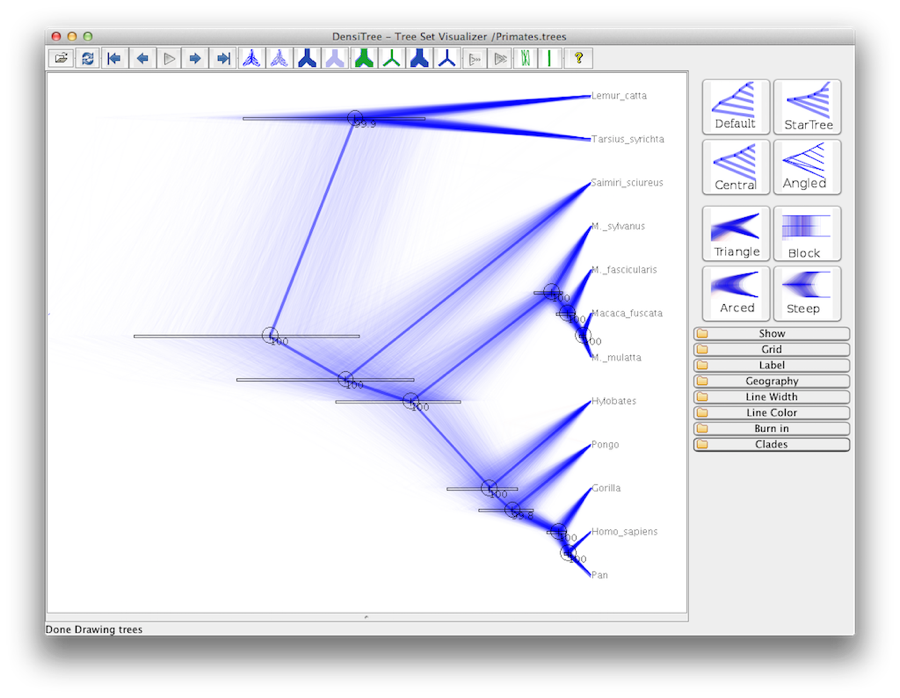
Abbildung 19: Ein Screenshot von FigTree und DensiTree.
Eine alternative Ansicht des Baums kann mit DensiTree erstellt werden, das Teil von Beast 2 ist. Der Vorteil von DensiTree besteht darin, dass es sowohl die Unsicherheit in den Knotenhöhen als auch die Unsicherheit in der Topologie visualisieren kann. Für diesen speziellen Datensatz ist die dominante Topologie in mehr als 99 % der Proben vorhanden. Daraus schließen wir, dass diese Analyse zu einem sehr hohen Konsens in Bezug auf die Topologie führt (Abbildung 19).
Fragen
- Unterscheidet sich die Evolutionsrate zwischen den verschiedenen Linien im Baum erheblich?
- DensiTree verfügt über eine Kladenleiste (Menüfenster/Symbolleiste Kladen anzeigen), um Informationen über Kladen anzuzeigen.
Wie hoch ist die Unterstützung für die Klade?
- Sie können die Topologien in DensiTree mit dem Menü Durchsuchen durchsuchen.Die beliebteste Topologie hat eine Unterstützung von über 99 %.
Wie hoch ist die Unterstützung für die zweitbeliebteste Topologie?
- Unter dem Hilfemenü zeigt DensiTree einige Informationen an.
Wie viele Topologien sind im Baumsatz?
8 Vergleich der Ergebnisse mit dem Prior
Es ist eine gute Idee, die Analyse zu wiederholen und dabei Stichproben aus dem Prior zu nehmen, um sicherzustellen, dass die Wechselwirkungen zwischen den Priors die Prior-Informationen nicht beeinträchtigen. Die Interaktion zwischen den Prioren kann vor allem bei der Verwendung von Kalibrierungen problematisch sein, da dies bedeutet, dass der Baum mit mehreren Prioren versehen wird.
Verwenden Sie BEAUti, um dieselbe Analyse zu erstellen, aber wählen Sie unter den MCMC-Optionen die Option „Sample from prior only“. Dadurch können Sie die vollständige Prior-Verteilung in Abwesenheit Ihrer Sequenzdaten visualisieren. Fassen Sie die Bäume aus der vollständigen Prior-Verteilung zusammen und vergleichen Sie die Zusammenfassung mit dem Posterior-Zusammenfassungsbaum.
Die Schätzung der Divergenzzeit mit Hilfe der „Knoten-Datierung“, wie sie in diesem Kapitel beschrieben wird, wurde zur Beantwortung einer Vielzahl verschiedener Fragen in der Ökologie und Evolution eingesetzt. Zum Beispiel wurde die Knoten-Datierung mit Fossilien bei der Bestimmung der Artenvielfalt von Cycaden, der Analyse der Evolutionsrate von Blütenpflanzen und der Untersuchung der Ursprünge von Cyanobakterien in heißen und kalten Wüsten verwendet.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond und Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), no. 1, 114. A Gelman, G Roberts, and W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled und Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, and S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian und Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith und Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008), no. 5898, 86-89.
Dieses Dokument wurde vonHEVEA aus LATEX übersetzt.