Einführung
De novo Mutationen in der Keimbahn (DNMs) sind genetische Veränderungen im Individuum, die durch Mutagenese in den elterlichen Gameten während der Oogenese und Spermatogenese entstehen. Dabei ist der Begriff „de novo“ nicht mit dem Begriff „neuartige Mutation“ zu verwechseln. Obwohl DNMs im Kontext eines Trios (Vater, Mutter und Kind) neuartige Mutationen sind, können sie in der allgemeinen Bevölkerung häufig, selten oder neuartige Varianten sein. Um die Häufigkeit einer bestimmten DNM zu messen und zu erklären, ist es notwendig, zunächst die Auswirkungen der Variante auf den Phänotyp zu bewerten, da sich neue vorteilhafte Merkmale entwickeln können, wenn auftretende genetische Mutationen einen spezifischen Überlebensvorteil bieten (Front Line Genomics, 2017).
Beim Menschen mit genetischen nicht-mendelschen Krankheiten, die sporadisch auftreten, sind DNMs in der Regel neu, zuverlässiger und schädlicher als vererbte Varianten, da sie keiner starken natürlichen Selektion unterliegen (Crow, 2000; Front Line Genomics, 2017). Daher kann es aus klinischer Sicht eine Herausforderung sein, die genetische Ursache einer durch eine DNM ausgelösten Störung bei einer Person zu identifizieren, da einem einzigen Phänotyp eine Pleiotropie und genetische Heterogenität zugrunde liegen kann (Eyre-Walker und Keightley, 2007). Dementsprechend wurden in den letzten zehn Jahren erhebliche Anstrengungen unternommen, um Exome von Personen mit Krankheiten unklarer genetischer Ätiologie zum Zwecke der klinischen Diagnostik zu sequenzieren. Doch selbst nach der Entdeckung von De-novo-Kandidatenvarianten gibt es immer noch unzureichende Informationen über die häufigen und seltenen Varianten, was eine klare Schlussfolgerung über die Pathogenität der identifizierten De-novo-Variante und ihre Rolle bei der Krankheit verhindert (Acuna-Hidalgo et al., 2016). Diese Einschränkung lässt sich dadurch erklären, dass De-novo-Varianten in der Regel heterozygot sind und entweder extrem selten oder häufig vorkommen können. Bei sehr seltenen De-novo-Varianten kann die Pathogenität der Variante schwer zu beweisen sein, da es keine weiteren Patienten mit demselben Phänotyp und derselben De-novo-Variante gibt. Bei häufigen de novo-Varianten sind die Faktoren, die für die Pathogenität der Variante ausschlaggebend sind, möglicherweise nicht bekannt, insbesondere wenn einige Personen in der Allgemeinbevölkerung die Variante haben, aber nicht an der genetischen Krankheit leiden. Unabhängig von der Rate der de novo-Varianten können jedoch beide Arten von Varianten auf der Grundlage der relativen Fitness und der natürlichen Selektion skaliert werden.
Die Anpassungsfähigkeit hängt von vielen Faktoren ab; um zu beurteilen, ob eine DNM pathogen oder adaptiv ist, und um zu verstehen, warum sie mit einer bestimmten Häufigkeit in der Population auftritt, ist es daher notwendig, die Variante unter geeigneten Bedingungen zu untersuchen. Dazu gehören die Umwelt, das Alter der Eltern, der genomische Kontext, die Epigenetik und andere Faktoren, da sie alle den Wert der mittleren relativen Fitness beeinflussen, der monoton ansteigt, während die Stärke der Selektion abnimmt (Peck und Waxman, 2018).
Das Hauptziel dieser Studie war es, die Häufigkeit des Auftretens von DNMs zu klären und zu bestimmen, wie diese Mutationen in den Exomen der allgemeinen litauischen Bevölkerung verteilt sind. Wir untersuchten auch, ob die Häufigkeit dieser Mutationen von der Zusammensetzung oder den strukturellen Parametern der Sequenzen, in denen sie auftraten, sowie von anderen Faktoren beeinflusst wurde, die die Mechanismen der Bildung dieser DNMs beeinflussen könnten. Schließlich wollten wir feststellen, ob DNMs aufgrund des intensiven Drucks der natürlichen Selektion auf die funktionellen Regionen entstanden sind. Obwohl die Verteilung und Intensität von DNMs Gegenstand vieler Studien waren, wurden sie in der litauischen Bevölkerung bisher nicht untersucht.
Materialien und Methoden
In dieser Studie analysierten wir Proben aus der litauischen Bevölkerung, die aus dem LITGEN-Projekt (LITGEN, 2011) stammen. Der Datensatz bestand aus 49 Trios mit insgesamt 144 verschiedenen Individuen. Genomische DNA wurde aus venösem Blut entweder mit der Phenol-Chloroform-Extraktionsmethode oder mit der automatisierten DNA-Extraktionsplattform TECAN Freedom EVO® (Tecan Schweiz AG, Schweiz) auf der Grundlage der paramagnetischen Partikelmethode extrahiert. Die Exome wurden auf einem SOLiD 5500 Sequenziersystem sequenziert (75 bp reads). Die Sequenzierdaten wurden mit der Lifescope-Software verarbeitet und aufbereitet. Die Exome wurden entsprechend dem menschlichen Referenzgenom Build 19 kartiert. Die durchschnittliche Lesetiefe der Sequenzierung betrug 38,5. BAM-formatierte Dateien von Mutter, Vater und Kind, die von Lifescope generiert wurden, wurden mit der SAMtools-Software für jedes Trio kombiniert.
De-novo-Mutationen wurden mit zwei Softwareprogrammen identifiziert: VarScan (Koboldt et al., 2012) und VarSeqTM. Eine potenzielle Variante wurde als DNM eingestuft, wenn sie in den Nachkommen identifiziert wurde, aber in keinem der Elternteile an der gleichen Position vorhanden war. Insgesamt wurden 1.752 und 4.756 DNMs durch VarScan bzw. VarSeqTM entdeckt. Um falsch-positive De-novo-Calls auszuschließen, bei denen nicht bekannt war, ob alle Individuen des Trios korrekt identifiziert wurden, wurden konservative Filter auf die erkannten DNM-Qualitätsparameter wie folgt angewendet: (1) Genotyp-Qualität des Individuums ≥50; (2) Anzahl der Reads an jeder Stelle >20. Die Software SnpSift wurde verwendet, um diese Filter auf die von VarScan generierten Daten anzuwenden. Die von der VarSeqTM-Software erzeugten Daten wurden gefiltert, indem dieselben Filterparameter im Segment Trio Workflow ausgewählt wurden. Um die verbleibenden somatischen Varianten (nur in einem Bruchteil der sequenzierten Blutzellen vorhanden) mit geringer Allelbalance oder Sequenzierungsartefakten auszuschließen, wurden DNMs gefiltert, indem ein Schwellenwert für den beobachteten Anteil der Reads in Individuen mit dem alternativen Allel (die Allelbalance) für das Trio festgelegt wurde (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Darüber hinaus wurden alle möglichen identifizierten und gefilterten de novo Einzelnukleotidvarianten manuell mit Integrative Genomics Viewer (Robinson et al., 2011) überprüft. Aufgrund der großen Anzahl identifizierter DNMs wurden für die Validierung von Varianten durch Sanger-Sequenzierung 51 de novo Einzelnukleotidvarianten zufällig ausgewählt. Die Sanger-Sequenzierung wurde mit einem ABI PRISM 3130xl Genetic Analyzer durchgeführt. Alle gefilterten und manuell überprüften DNMs, die durch VarScan (N = 95) und VarSeqTM (N = 84) identifiziert wurden, wurden mit ANNOVAR (Butkiewicz und Bush, 2016; Wang et al., 2010) annotiert. Für die Analyse der Proteininteraktionen wurde die Software STRING (Szklarczyk et al., 2017) verwendet. Wie bei der Exomkartierung wurden die Annotationen anhand des menschlichen Referenzgenoms hg19 durchgeführt.
Die Wahrscheinlichkeit, dass eine aufgerufene Position eine DNM im Trio war, wurde unabhängig für jedes Trio berechnet. Wie in einer früheren Referenz beschrieben (Besenbacher et al., 2015) beschrieben, wurde die De-novo-Rate pro Position pro Generation (PPPG) wie folgt berechnet:
wobei f die Anzahl der Trios und N die Anzahl der Callable Sites ist, die potenziell als De-novo-Sites für jedes Trio separat identifiziert werden können, unabhängig von der Sequenziertiefe. Diese Zahl variiert je nach Trio. ni ist die Zahl der identifizierten DNMs für Trio i. Die Wahrscheinlichkeit Pji (de novos ingle nucleotide) für die genannte einzelne Nukleotidstelle j und Familie i, mutiert zu sein, wurde wie folgt berechnet:
Die Wahrscheinlichkeit Pji (de novo indel)für die genannte Indel-Stelle j und Familie i, mutiert zu sein, wurde berechnet als:
wobei C, M und F für Nachkommen, Mutter bzw. Vater stehen und Hetero, HomR und HomA heterozygot, homozygot für Referenz bzw. homozygot für alternatives Allel bedeuten. Die Wahrscheinlichkeit Pij (de novo) wurde in Abhängigkeit von der Sequenzierabdeckung berechnet. Konfidenzintervalle für Ratenschätzungen wurden wie für binomiale Proportionen berechnet. Für die Schätzung der DNM-Rate und für weitere Berechnungen verwendeten wir das R-Paket (Version 3.4.3) (R Core Team, 2013).
Um die Hypothese zu testen, dass Variationen in der DNM-Rate in verschiedenen Regionen des Genoms durch intrinsische Eigenschaften der genomischen Region selbst und das Alter der Eltern erklärt werden könnten, wurde eine lineare Regressionsanalyse durchgeführt, für die die „sekundäre“ Annotation jeder DNM unter Verwendung von Daten aus den Projekten ENCODE (ENCODE Project Consortium, 2012) und LITGEN (LITGEN, 2011) durchgeführt wurde. Zunächst wurden gemäß einer früheren Studie (Besenbacher et al., 2015) Lymphoblastoid-Zelllinien (LCL und GM12878) (ENCODE Project Consortium, 2012) ausgewählt, um Daten über die genomische Landschaft der identifizierten DNMs zu sammeln. Es wurden Daten für:
(1) Expressionsraten (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) in verschiedenen Geweben gesammelt. Entsprechend der Expression von Regionen mit DNMs wurden in Positionen mit spezifischer und unspezifischer Expression unterteilt;
(2) Messungen von DNase1-Hypersensitivitätsstellen (DHS). Der DHS-Status wurde mit 0 bewertet, wenn er außerhalb des DHS-Peaks lag, und mit 1, wenn er innerhalb lag;
(3) Messungen des Kontexts der CpG-Inseln. Befand sich die DNM innerhalb der CpG-Inseln, wurde ihr der Status 1 zugewiesen; lag sie außerhalb, 0;
(4) drei Histonmarkierungen (H3K27ac, H3K4me1 und H3K4me3) aus dem ENCODE-Projekt. Wenn DNM in einer mit Histon markierten Position war, wurde sie mit 1 bewertet, wenn nicht, mit 0;
(5) GERPP++ Erhaltungswerte wurden mit dem Annotationstool ANNOVAR erfasst. Entsprechend der Erhaltungswerte wurden Positionen mit DNMs in konservative (GERP++ Score >12) und nicht konservative Positionen (GERP++ Score <12) eingeteilt (Davydov et al., 2010; ENCODE Project Consortium, 2012). Auf der Grundlage von Fragebogendatensätzen aus dem LITGEN-Projekt wurden Daten zum Alter der Eltern gesammelt. Nach der Erfassung der Parameter für jedes Trio wurde eine Anzahl von Positionen mit jedem Parameter berechnet. Dann wurde eine Korrelationsanalyse mit anschließender linearer Regressionsmodellierung der DNM-Rate und der Parameter durchgeführt.
Ergebnisse
Nach der DNM-Analyse wurde für zwei Trios (Nr. 4 und 21) eine außergewöhnlich hohe Anzahl von DNMs identifiziert: 113 und 123 (durch VarScan bzw. VarSeqTM) und 16 (VarScan). Diese Ergebnisse veranlassten uns, die biologische Vaterschaft zu testen, die für Trio Nr. 4 abgelehnt und für Trio Nr. 21 bestätigt wurde. 21. Daher wurden die Daten des Trios Nr. 4 aus der Studie ausgeschlossen. In der endgültigen Gruppe von 48 Trios wurden 95 DNMs in 34 Trios mit der VarScan-Software und 84 DNMs in 31 Trios mit der VarSeqTM-Software identifiziert (Abbildung 1). In 18 bzw. 15 Trios wurden mit VarScan bzw. VarSeqTM keine DNMs entdeckt. Von allen DNMs, die von beiden Softwareprogrammen identifiziert wurden, stimmten nur 5,37 % der DNMs überein (drei DNMs in den Genen MEIS2, PGK1 und MT1B). Jede Person hatte im Durchschnitt 1,9 (VarScan-Software) und 1,7 (VarSeqTM) DNMs.
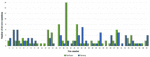
ABBILD 1. Vergleich der von VarScan (blau) und VarSeqTM (grün) identifizierten de novo Einzelnukleotidvarianten.
Die Analyse von 95 DNMs, die von der VarScan-Software identifiziert wurden, zeigte, dass 20 DNMs exonisch waren, darunter zwei Stop-Gain-DNMs, sieben synonyme DNMs und 11 nicht-synonyme DNMs. Achtzig neue Mutationen, die mit VarSeqTM identifiziert wurden, waren exonisch, darunter 1 Stop-Gain-DNM und 78 nicht-synonyme DNMs (Abbildung 2). Die Mehrzahl der von VarScan identifizierten DNMs befand sich auf den Chromosomen 1, 2, 4 und 5, während VarSeqTM DNMs überwiegend auf den Chromosomen 2, 6, 7 und 11 identifizierte. Die Anzahl der identifizierten DNMs korrelierte weder mit der Dichte der Gene auf den Chromosomen (R = 0,09, p-Wert = 0,65 für VarScan und R = 6,73, p-Wert = 0,51 für VarSeqTM) noch mit der Chromosomengröße (Abbildung 3). Nach beiden Softwareprogrammen waren die Verhältnisse von Übergängen und Transversionen sehr ähnlich: 1,44 bzw. 1,47 (Abbildung 4). Es wurden jedoch Unterschiede in den Strukturen der Übergänge festgestellt. Insbesondere gab es unter den von VarScan identifizierten DNMs mehr G/T- und A/C-Änderungen, während unter den von VarSeqTM identifizierten DNMs mehr A/T- und G/C-Änderungen auftraten.
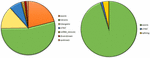
Abbildung 2. Die Zusammensetzung der De-novo-Mutationen (DNMs), die von VarScan (links) und VarSeqTM (rechts) erzeugt wurden.
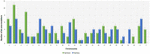
Abbildung 3. Verteilung der Anzahl der de novo-Varianten nach Chromosom gemäß den von VarScan und VarSeqTM generierten Daten. Grüne Balken stellen DNMs dar, die von der VarScan-Software identifiziert wurden, blaue – von VarSeqTM. Die Fehlerbalken stellen den Standardfehler der mittleren DNMs für jedes Chromosom dar.
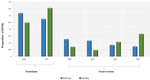
Abbildung 4. Die molekularen Ereignisse, die den Transitionen zugrunde liegen, treten häufiger auf als diejenigen, die zu Transversionen führen, was zu einer ∼1,5-fach höheren Rate von Transitionen gegenüber Transversionen im gesamten Exom führt. Mit der VarScan- (grün) und VarSeqTM-Software (blau) identifizierte Transitions- und Transversionsereignisse. Die Fehlerbalken stellen den Standardfehler der mittleren DNMs dar.
Die berechneten Raten der de novo Einzelnukleotidmutationen betrugen 2,4 × 10-8 PPPG (95% Konfidenzintervall: 1,96 × 10-8-2,99 × 10-8) gemäß VarSeqTM und 2.74 × 10-8 pro Nukleotid pro Generation (95% CI: 2,24 × 10-8-3,35 × 10-8) gemäß VarScan.
Drei de novo Indels in drei Trios wurden durch den VarScan-Algorithmus in den Chromosomen 6 und 11 identifiziert. Die berechnete Rate der de novo-Indels im Genom betrug 1,77 × 10-8 (95% CI: 6,03 × 10-9-5,2 × 10-8) PPPG. Bemerkenswert ist, dass alle De-novo-Indels „reversibel“ waren, d. h. die Eltern hatten neue Varianten im Genom, und ihre Kinder hatten De-novo-Varianten auf der Grundlage des Referenzgenoms mit dem 37,5-Mittelwert der Sequenziertiefe bzw. der 50-Genotyp-Qualität. Diese drei DNMs wurden jedoch nicht für die Validierung durch die Sanger-Sequenzierungsmethode ausgewählt, so dass die Wahrscheinlichkeit einer Überschätzung der De-novo-Indels dennoch bestehen bleibt. De-novo-Indels waren C/T und A/G im Kontext einzelner Nukleotide.
Die lineare Regressionsmodellierung ergab, dass DNAse-1-Hypersensitivitätsstellen, der Kontext von CpG-Inseln, GERPP++-Erhaltungswerte und Expressionsniveaus ∼68-93 % der DNM-Raten erklärten (Tabelle 1). Weder epigenetische Marker noch das väterliche Alter korrelierten signifikant mit der DNM-Rate. Die Modelle wurden nur aus den Daten von VarScan erstellt, da es keine Korrelation zwischen den Daten von VarSeqTM und den intrinsischen Merkmalen der genomischen Region selbst gab.

TABLE 1. Die lineare Regression der DNAaseI-Hypersensitivitätsstellen, des Kontexts der CpG-Inseln, der GERPP++-Erhaltungswerte und der Auswirkung des Expressionsniveaus auf die Rate der DNMs.
Funktionelle Vorhersage von DNMs
Um zu beurteilen, welche Fehlsensemutationen schädlich waren und die Funktion des betroffenen Proteins nach Typ veränderten, wurden vorhergesagte kategorische Scores für den durch DNMs induzierten Schaden analysiert. Die folgenden 10 Werte wurden berücksichtigt: Polyphen HDIV und HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, Fathmm-MKL coding und GERP++. Auf der Grundlage der vorhergesagten Scores wurden vier DNMs ausgewählt, die von VarScan als sechs oder mehr schädliche oder wahrscheinlich schädliche Vorhersagen identifiziert wurden. Diese Stop-Gain-DNMs befanden sich in den Genen MEIS2 und ULK4, während nicht-synonyme DNMs in den Genen MT1B und PGK1 auftraten. Die von diesen Genen kodierten Proteine sind wichtig für das neuronale Wachstum, die Endozytose und den Schutz vor den negativen Auswirkungen von Schwermetallen. Diese Proteine sind an der Freisetzung des Tumorblutgefäßhemmers Angiostatin und an verschiedenen Signalwegen beteiligt. Es gab keine Verbindungen zwischen den von diesen Genen kodierten Proteinen (Abbildung 5).
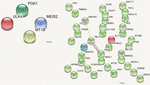
ABBILD 5. Protein-Protein-Interaktionen (Szklarczyk et al., 2017) in Genen, die DNMs beherbergen. Durch VarScan identifizierte DNMs in Genen, die Proteine kodieren, stehen links, durch VarSeqTM identifizierte DNMs rechts. Farbige Linien zeigen eine Verbindung zwischen Proteinen an.
De-novo-Mutationen, die von VarSeqTM identifiziert wurden, wurden genauer analysiert, wenn sie von mindestens der Hälfte der Vorhersage-Tools als schädlich oder wahrscheinlich schädlich vorhergesagt wurden. Es gab 35 Punktmutationen (siehe ??) in Genen, die für Proteine kodieren, die für den Umbau des Chromatins, die Regulierung des Zytoskeletts, das Zellwachstum und die Lebensfähigkeit, zytoplasmatische Signalwege und die Auslösung neuronaler Reaktionen, die die Geruchswahrnehmung auslösen, wichtig sind.
Unter den Proteinen, die von den DNM-beeinträchtigten Genen kodiert werden, waren nur CLPTM1, ZNF547 und DMXL1 in irgendeiner Weise miteinander verbunden (Abbildung 5).
Diskussion
In dieser Studie haben wir eine umfassende Analyse der Verteilung von DNMs über verschiedene Regionen des Exoms in der litauischen Bevölkerung durchgeführt. Insgesamt wurden 95 DNMs in 34 Trios und 84 DNMs in 31 Trios mit der SOLiD 5500 Sequenzierungstechnologie durch VarScan- bzw. VarSeqTM-Algorithmen nachgewiesen. Zunächst möchten wir anmerken, dass wir VarScan für das DNM-Calling gewählt haben, weil dieser Algorithmus laut (Warden et al., 2014) eine Liste von Varianten mit hoher Übereinstimmung (>97%) mit hochwertigen Varianten ergibt, die von GATK UnifiedGenotyper und HaplotypeCaller genannt werden. Die VarSeqTM-Software wurde ausgewählt, weil sie ein weit verbreitetes Werkzeug für die Variantenanalyse sowohl in der Forschung als auch in der klinischen Analyse ist. Obwohl beide Algorithmen darauf ausgelegt sind, im Exom der Nachkommen nach DNMs zu suchen, die in beiden Elternteilen nicht vorhanden waren, betrug die Übereinstimmung zwischen den beiden Softwareprogrammen für die DNM-Analyse nur 5,37 %. Der VarScan-Algorithmus hatte eine höhere Sensitivität (5,42 %) für die Erkennung von DNMs vor der Filtration als der VarSeqTM-Algorithmus (1,77 %), so dass wir vermuteten, dass kein Tool aufgrund der hohen Sensitivität, die immer mit einer niedrigen Spezifität einherging, erfolgreich Mutationen benennen konnte. Daher schlagen wir vor, dass eine erhebliche Verbesserung der Ergebnisse durch die Kombination der Ergebnisse verschiedener Tools erreicht werden könnte (Sandmann et al., 2017).
Basierend auf den generierten Daten lag die geschätzte Einzelnukleotid-DNM-Rate zwischen 2,4 × 10-8 und 2,74 × 10-8 und die von de novo Indels bei 1,77 × 10-8 PPPG, abhängig vom verwendeten Algorithmus. Die von uns berechnete DNM-Rate war höher als in früheren Studien (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015), in denen sie zwischen 1,2 × 10-8 und 1,5 × 10-8 PPPG lag. Die höhere DNM-Rate in unserer Studie war vertretbar, da unsere Studie auf Exom-Daten basierte. Außerdem weisen Exome deutlich höhere Mutationsraten (um 30 %) auf als ganze Genome, da sich die Basenpaar-Zusammensetzung des gesamten Genoms von derjenigen der Exome unterscheidet. Insbesondere haben Exome einen durchschnittlichen GC-Gehalt von etwa 50 %, während der GC-Gehalt des gesamten Genoms bei etwa 40 % liegt (Neale et al., 2012). Methylierte CpGs stellen beim Menschen aufgrund der spontanen Desaminierung von Cytosinbasen stark veränderliche Sequenzen dar (Neale et al., 2012). Vergleichenden Genomikstudien zufolge ist davon auszugehen, dass sich die erhöhten Mutationsraten in CpG-reichen Regionen um die Zeit der Radiation der Säugetiere entwickelt haben (Francioli et al., 2015). Während der Divergenz der Spezies unterlagen CpG-reiche exonische Regionen im Vergleich zu denen an nicht codierender DNA erhöhten Mutationsraten und wurden zu nicht codierenden Regionen. Daher nimmt die Wirkung des CpG-Gehalts im Laufe der Zeit ab und die durchschnittliche Mutationsrate sinkt, bis sie das Niveau der umgebenden nicht-kodierenden DNA erreicht (Subramanian und Kumar, 2003). Während jedoch Sequenzen in sich neutral entwickelnden Regionen des Genoms ausreichend Zeit hatten, sich in Bezug auf Dinukleotidkontexte auszugleichen, hat die reinigende Selektion hypermutable CpGs in funktionalen Regionen aufrechterhalten (Subramanian und Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Da wir eine höhere DNM-Rate als die in anderen Studien berichteten gefunden haben, spekulierten wir, dass dies zumindest teilweise auf den lokalen Sequenzkontext und/oder einen möglichen natürlichen Selektionsdruck auf das Exom zurückzuführen sein könnte. Dementsprechend wurde ein lineares Regressionsmodell angewandt, und wir fanden heraus, dass die DNAse-1-Hypersensitivität, der Kontext der CpG-Inseln, die GERPP++-Erhaltungswerte und das Expressionsniveau ∼68-93 % der DNM-Rate erklärten. Diese Ergebnisse deuten darauf hin, dass sich DNMs im Exom unabhängig von der Erhaltung von DNA-Sequenzen bilden. Allerdings war die DNM-Rate bei Genen, deren Produkte unspezifisch waren, und in transkriptionell aktiven Promotor-ähnlichen Regionen höher.
Im Gegensatz zu den Ergebnissen anderer Studien (Wong et al., 2016; Sandmann et al., 2017) fanden wir, dass das väterliche Alter nicht mit der DNM-Rate korrelierte. Diese Ergebnisse könnten dadurch erklärt werden, dass der Datensatz aus Trios mit ähnlichem Alter der Eltern bestand und dass nur ein kleiner Teil (∼1,5 %) des gesamten Genoms analysiert wurde. Basierend auf diesen Parametern hatte jede Person im Durchschnitt nur 1,9 (VarScan) bzw. 1,7 (VarSeqTM) DNMs im Vergleich zu 40-82 im gesamten Genom (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al, 2016), während die Anzahl der De-novo-Indels in der kodierenden Sequenz ähnlich war wie in (Front Line Genomics, 2017).
Die Ergebnisse unserer umfassenden funktionellen Analyse der Annotationen zeigten, dass von allen identifizierten DNMs 4 (VarScan) und 35 (VarSeqTM) Varianten wahrscheinlich pathogene DNMs waren. Der Unterschied in der Anzahl der pathogenen DNMs lässt sich möglicherweise dadurch erklären, dass sich der Anteil der DNMs in kodierenden Sequenzen je nach dem für die Identifizierung von DNMs verwendeten Algorithmus erheblich unterscheidet. So waren beispielsweise 21,05 % der von der VarScan-Software identifizierten DNMs exonisch, während 95,24 % der von der VarSeqTM-Software identifizierten DNMs exonisch waren. Diese pathogenen DNMs befanden sich in den Genen, die für Proteine kodieren, die für die Chromatinmodellierung, die Regulierung des Zytoskeletts, die Modulation des Zellwachstums und der Vitalität, die Funktion zytoplasmatischer Signalwege und die Initiierung neuronaler Reaktionen wesentlich sind. Obwohl diese DNMs als pathogen angesehen werden, bezeichneten sich alle an der Umfrage teilnehmenden Personen als genetisch „gesund“. Dieses Ergebnis deutet also darauf hin, dass trotz der vermuteten Pathogenität der DNMs die Genome, in denen sich die DNMs befanden, solche Veränderungen offensichtlich tolerierten, so dass die Krankheitsmanifestationen oft nicht ausgeprägt waren. Nach Szamecz et al. (2014) sind die Auswirkungen der natürlichen Selektion auf genetische Veränderungen durch kompensatorische Mechanismen des Genomschutzes umso stärker, je häufiger DNMs an konservierten Genpositionen auftreten. Die schädlichen Auswirkungen der Varianten können auf vier Arten abgeschwächt werden. Einige Gene können verkürzte Varianten von Proteinen tolerieren, weil ihre funktionellen Auswirkungen durch eine unvollständige Expression, kompensatorische Varianten oder eine geringe funktionelle Bedeutung der Verkürzung maskiert werden (Bartha et al., 2015). Im Gegensatz dazu werden Genveränderungen, die mit nicht-synonymen DNMs verbunden sind, durch den Mechanismus der nützlichen Mutationsakkumulation im gesamten Genom kompensiert (Szamecz et al., 2014). Dies deutet darauf hin, dass in diesen Fällen die pathogenen Mutationen nicht schädlich genug sind, um die durchschnittliche Fitness zu verringern, und dass sie daher über viele Generationen hinweg bestehen bleiben und durch natürliche Selektion geformt werden.
Zusammenfassend lässt sich sagen, dass unsere Analyse der Verteilung von DNMs und ihres genetischen und epigenetischen Kontexts Einblicke in die genetische Variation des litauischen Genoms lieferte. Auf der Grundlage dieser Ergebnisse könnten zusätzliche Studien an Patientengruppen mit genetischen Erkrankungen unsere Fähigkeit verbessern, bestimmte pathogene DNMs von den tolerierten Hintergrund-DNMs zu unterscheiden und zuverlässige ursächliche DNMs zu identifizieren. Die wichtigste Einschränkung dieser Studie bestand jedoch darin, dass wir die Variation in nicht kodierenden und regulatorischen Genregionen nicht untersucht haben. Diese Informationen könnten zur Aufklärung möglicher Mechanismen der DNM-Bildung beitragen, die noch immer nicht ausreichend geklärt sind.
Accession Codes
Die Sequenzdaten wurden im European Nucleotide Archive (ENA) unter dem Zugang PRJEB25864 (ERP107829) hinterlegt.
Ethikerklärung
Diese Studie wurde in Übereinstimmung mit den Empfehlungen der regionalen Ethikkommission für biomedizinische Forschung in Vilnius durchgeführt. Das Protokoll wurde von der regionalen Ethikkommission für biomedizinische Forschung in Vilnius genehmigt. Alle Probanden gaben ihre schriftliche Einwilligung gemäß der Deklaration von Helsinki.
Beiträge der Autoren
LP führte die Datenanalyse durch und verfasste das Manuskript. AJ berechnete die Rate der De-novo-Mutationen. Die Sequenzierung von Trios Exomen wurde von LA und IK durchgeführt. VK war der Hauptforscher.
Finanzierung
Diese Studie wurde vom Europäischen Sozialfonds im Rahmen der Global Grant Maßnahme unterstützt. LITGEN Projekt Nr.. VP1-3.1-ŠMM-07-K-01-013.
Erklärung zu Interessenkonflikten
Die Autoren erklären, dass die Forschung in Abwesenheit jeglicher kommerzieller oder finanzieller Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Ergänzendes Material
Das ergänzende Material zu diesem Artikel finden Sie online unter: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). Neue Einblicke in die Entstehung und Rolle von De-novo-Mutationen in Gesundheit und Krankheit. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). Die Eigenschaften von heterozygoten Proteinabbruchvarianten im menschlichen Genom. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Neue Variation und De-novo-Mutationsraten in populationsweit de novo assemblierten dänischen Trios. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). CpG-Insel-Cluster und pro-epigenetische Selektion für CpGs in proteincodierenden Exons von HOX und anderen Transkriptionsfaktoren. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). In silico functional annotation of genomic variation. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Die Ursprünge, Muster und Auswirkungen der menschlichen Spontanmutation. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., und Batzoglou, S. (2010). Identifizierung eines hohen Anteils des menschlichen Genoms, der unter Selektionszwang steht, mit GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Eine integrierte Enzyklopädie der DNA-Elemente im menschlichen Genom. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., and Keightley, P. D. (2007). Die Verteilung der Fitnesseffekte neuer Mutationen. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genomweite Muster und Eigenschaften von De-novo-Mutationen beim Menschen. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics Magazine Issue 14 – ASHG. London: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Genetische Effekte auf die Genexpression in menschlichen Geweben. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: Entdeckung somatischer Mutationen und Kopienzahlveränderungen bei Krebs durch Exom-Sequenzierung. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate der de novo Mutationen und die Bedeutung des Alters des Vaters für das Krankheitsrisiko. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Feinräumige Unterschiede in der Rekombinationsrate zwischen Geschlechtern, Populationen und Individuen. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ‚t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Transkriptom- und Genomsequenzierung deckt funktionelle Variation beim Menschen auf. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Available at: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Muster und Raten von exonischen de novo Mutationen bei Autismus-Spektrum-Störungen. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Was ist Anpassung und wie sollte sie gemessen werden? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrativer Genomik-Viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., and Sunyaev, S. (2008). Hypermutable nicht-synonyme Stellen sind einer stärkeren negativen Selektion ausgesetzt. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Neutrale Substitutionen treten in Exons schneller auf als in nichtcodierender DNA in Primatengenomen. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). Die genomische Landschaft der kompensatorischen Evolution Be. Die genomische Landschaft der kompensatorischen Evolution. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). Die STRING-Datenbank im Jahr 2017: Qualitätskontrollierte Protein-Protein-Assoziationsnetzwerke, breit zugänglich gemacht. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: funktionale Annotation von genetischen Varianten aus Next-Generation-Sequencing-Daten. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Detaillierter Vergleich von zwei populären Variantenaufruf-Paketen für Exom- und gezielte Exon-Studien. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Neue Beobachtungen zum Einfluss des mütterlichen Alters auf Keimbahn-De-novo-Mutationen. Nature Communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar