Inzidenz und Prävalenz von Nierensteinerkrankungen nehmen in der Allgemeinbevölkerung weiter zu. Mit einer Lebenszeitprävalenz von bis zu 10 % stellen Nephrolithiasis (NL) und Nephrokalzinose (NC) daher eine große Gesundheitsbelastung dar, insbesondere in der westlichen Welt (1). NL und NC sind mit einer erheblichen Morbidität und dem Fortschreiten einer chronischen Nierenerkrankung verbunden, was auf das Wiederauftreten, wiederholte chirurgische/endoskopische Eingriffe und begleitende Entzündungen zurückzuführen ist. Vereinfacht ausgedrückt, ist die Nierensteinbildung das Ergebnis eines Ungleichgewichts zwischen den Inhibitoren (z. B. Citrat, Magnesium, Uromodulin und Pyrophosphat) und den Promotoren (z. B. Oxalat, Kalzium, Phosphat, Urat und Cystin) der Kristallisation im Urin, das zu einer Übersättigung mit konsekutiver Aggregation, Keimbildung und Steinwachstum an der Randall’schen Plaque führt (Abbildung 1). Dieses Ungleichgewicht kann auf eine veränderte enterale und/oder renale Handhabung von Promotoren oder Inhibitoren zurückzuführen sein, wie z. B. eine enterale Fehlsekretion von Oxalat oder eine renale Malreabsorption von Kalzium (Abbildung 1).

Abbildung 1. Ungleichgewicht von Harninhibitoren und Promotoren der Kristallisation, das zur Nierensteinbildung führt. Die Konzentration von Inhibitoren und Promotoren im Urin wird sowohl von intestinalen als auch renalen Transportern beeinflusst und kontrolliert (GI-Nieren-Achse). Diese Transporter und Austauscher, wie SLC26A1, sind für die Sekretion und Absorption verantwortlich. In Bezug auf Oxalat führt eine enterale Hyperabsorption, aber eine Malsekretion und/oder eine renale Hypersekretion, aber eine Malabsorption dazu, dass die Oxalatkonzentration im Urin die Übersättigung übersteigt und dadurch die Kristallisation und die nachfolgende Steinbildung begünstigt wird.
Man geht davon aus, dass die zugrundeliegende Ätiologie der NL multifaktoriell ist und eine umweltbedingte, ernährungsbedingte, hormonelle und genetische Komponente aufweist. In Zwillingsstudien wurde die Erblichkeit von Nierensteinen auf 56 % geschätzt (3), und bis zu zwei Drittel der hypercalciurischen Steinbildner haben Verwandte mit NL (4). Obwohl mehr als 80 % aller Nierensteine kalziumhaltig sind, ist die genetische Grundlage dieser Steine noch weitgehend unbekannt (5). Mit Ausnahme von Varianten in CLDN14, TRPV5, SLC34A1, ALPL, CASR und UMOD haben genomweite Assoziationsstudien noch keine wesentlichen genetischen Faktoren ergeben (6-8). Es wurden jedoch Risikoallele innerhalb von Genen identifiziert, die die Krankheit auch auf mendelscher Basis übertragen, wie CASR, SLC34A1 und SLC2A9 (9, 10). Bis heute wurden mehr als 30 einzelne Gene mit einem Online Mendelian Inheritance in Man definierten Phänotyp identifiziert, die, wenn sie mutiert sind, an NL/NC beteiligt sind (Tabelle 1).

Table 1. Gene, von denen bekannt ist, dass sie monogene Formen von NL/NC verursachen.
Die Vererbungsmodi bei monogenen Formen umfassen autosomal-dominante, autosomal-rezessive und X-chromosomale Übertragung. Interessanterweise wurde bei mehreren dieser Gene sowohl ein rezessiver als auch ein dominanter Vererbungsmodus festgestellt: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 und SLC4A1. Während die meisten syndromalen und schweren kongenitalen Störungen einen rezessiven Erbgang aufweisen (Bartter, Lowe, Dent, FHHNC und distale renale tubuläre Azidose mit sensorineuraler Taubheit), sind mildere Erkrankungen eher mit Mutationen in dominanten Genen verbunden. Bei der Mehrzahl der kodierten Proteine handelt es sich um renale Transporter für gelöste Stoffe (z. B. SLC34A1, SLC34A3 und SLC9A3R1), aber auch Chloridkanäle (CLCN5), Tight-Junction-Proteine (z. B. CLDN16/CLDN19) und metabolisierende Enzyme (z. B. AGXT, APRT und CYP24A1) sind bei Patienten mit NL/NC defekt. Der zugrundeliegende Defekt ist daher meist im Tubulussystem der Niere selbst zu finden und kann daher als Tubulopathie bezeichnet werden. Umgekehrt sind a priori extrarenale Bedingungen wie bei der primären Hyperoxalurie (PH), bei der eine Funktionsstörung von Leberenzymen (AGXT, GRHPR und HOGA1) eine Oxalatakkumulation mit sekundärer Nierenschädigung verursacht, als Ursachen für NL/NC denkbar. Obwohl man davon ausgeht, dass jeder Krankheitsphänotyp eine relativ seltene Entität darstellt, können die Ursachen für ein einzelnes Gen aufgrund ihrer großen genetischen Heterogenität eine beträchtliche Anzahl von Patienten ausmachen (42). Neben der genetischen Heterogenität gibt es auch eine allelische Variation, wobei trunkierende Varianten eher zu einem Funktionsverlust führen und Missense-Varianten (Hypomorphe) eher subtile Defekte verursachen können, die vor allem bei erwachsenen Steinbildnern klinisch übersehen werden können. Ein weiteres, erst kürzlich bekannt gewordenes Phänomen sind Gendosierungseffekte in mehreren der oben genannten Nierensteingene. Bei SLC34A3 beispielsweise, das für einen der wichtigsten Phosphattransporter im proximalen Tubulus (NaPiIIc) kodiert, konnte gezeigt werden, dass heterozygote Individuen nicht mehr nur als gesunde Träger angesehen werden können, da sie signifikant häufiger Nierenverkalkungen und/oder Knochenmanifestationen aufweisen als Wildtyp-Individuen; allerdings immer noch in geringerem Maße als biallelische (homozygote und compound heterozygote) Individuen (43). Ähnliche Beobachtungen wurden für Familien mit Mutationen in CYP24A1 gemeldet (44). Der Beitrag monogener Störungen zur Gesamtprävalenz der Nierensteinerkrankung wurde in der Vergangenheit nicht umfassend untersucht. Insbesondere fehlt es an genetischen Erkenntnissen, die auf breit angelegten Untersuchungen einer Vielzahl von ursächlichen Genen in großen Patientenkohorten beruhen. Umfassende Gentests waren in der Vergangenheit zu kostspielig und ineffizient. Für die meisten Personen mit NL/NC war eine Mutationsanalyse für einen ursächlichen Gendefekt daher nicht zugänglich, obwohl die Kenntnis der molekularen Ursache von NL/NC wichtige Konsequenzen für Prognose, Prophylaxe und/oder Behandlung haben kann. Aus klinischen Beobachtungsstudien wurden nur grobe Schätzungen abgeleitet: Auf der Grundlage einer umfangreichen Datensammlung zur Analyse der Steinzusammensetzung kam man zu dem Schluss, dass monogene Ursachen bei Kindern nicht mehr als 9,6 % und bei Erwachsenen 1,6 % ausmachen (45). In den letzten zehn Jahren hat sich diese Situation jedoch mit dem Aufkommen von Hochdurchsatz-Sequenzierungstechniken zu ändern begonnen.
Hochdurchsatz-Mutationsanalyse bei Patienten mit NL/NC
Um Patienten mit Nierensteinleiden auf das Vorhandensein pathogener Mutationen in bekannten Krankheitsgenen zu untersuchen, haben wir ein Gen-Panel auf der Grundlage von mikrofluidischer Multiplex-PCR und konsekutiver NextGen-Sequenzierung (Fluidigm™/NGS) erstellt (46, 47).
In einer „Pilotstudie“ rekrutierten wir nacheinander 268 genetisch unauffällige Personen aus typischen Nierensteinkliniken; 102 pädiatrische und 166 erwachsene Probanden. Als Ergebnis identifizierten wir 50 schädliche Varianten in 14 von 30 analysierten Genen, was in 15 % aller Fälle zu einer molekularen Diagnose führte. In der pädiatrischen Untergruppe wiesen wir in 21 % eine ursächliche Mutation nach, während bei den Erwachsenen in 11 % schädliche Varianten vorhanden waren (Abbildung 2A) (48). Mutationen im Zystinurie-Gen SLC7A9 wurden am häufigsten in der Erwachsenenkohorte gefunden (Abbildung 2B). Zwei Folgestudien konnten diese Ergebnisse bestätigen. Erstens konnten in einer ausschließlich pädiatrischen Kohorte von 143 NL/NC-Patienten 17 % der Fälle durch Mutationen in 14 verschiedenen Genen erklärt werden (49). Zweitens konnte in einer Kohorte von 51 Familien mit einem Manifestationsalter der NL/NC von vor 25 Jahren durch gezielte WES in fast 30 % der Fälle eine genetische Ursache nachgewiesen werden (50). Es überrascht nicht, dass rezessive Mutationen häufiger bei Neugeborenen und bei angeborenen Erkrankungen gefunden wurden, während sich dominante Erkrankungen in der Regel später im Leben manifestieren. Diese Daten deuten darauf hin, dass genetisch bedingte Nierensteinleiden unterdiagnostiziert sind, obwohl die molekulare Diagnose die Prognose, Prophylaxe und/oder Behandlung beeinflussen kann. Eine erwähnenswerte Einschränkung ist jedoch eine mögliche Selektionsverzerrung aufgrund der Rekrutierung aus spezialisierten Nierensteinkliniken in allen drei vorgenannten Studien.

Abbildung 2. Mutationsanalyse von 30 bekannten monogenen Nephrolithiasis (NL)/Nephrocalcinose (NC)-Genen bei 268 Patienten mit NL/NC. (A) Anteil der monogenen Ursachen in der pädiatrischen und erwachsenen Subkohorte. (B) Anzahl der monogenen Ursachen bei allen Genen (rot bedeutet Erwachsene; orange bedeutet pädiatrische Patienten). Bemerkenswert ist, dass SLC7A9 am häufigsten mutiert war, vor allem bei jungen Erwachsenen.
Identifizierung neuer menschlicher Krankheitsgene durch Kandidatengen-Ansatz
Die Hochdurchsatz-Mutationsanalyse wird auch zur Suche nach pathogenen Varianten in verschiedenen Kandidatengenen eingesetzt. Eine der interessantesten jüngsten Entdeckungen war die Entdeckung menschlicher Mutationen in SLC26A1 (32). Seit der ersten Beschreibung der Ca-Oxalat (CaOx)-Nierensteinbildung und NC in Slc26a1 (Sat1)-Knockout-Mäusen durch Dawson et al. im Jahr 2010, ist SLC26A1 ein echtes NL-Kandidatengen (51). SLC26A1 kodiert für einen Anionenaustauscher, der an der basolateralen Membran von proximalen Nierentubuli, Ileum und Jejunum exprimiert wird. Mithilfe eines Kandidatengen-Ansatzes wurden pathogene Varianten bei Menschen mit einer Vorgeschichte von früh einsetzender CaOx-NL identifiziert, und zwar bei zwei nicht verwandten Personen mit biallelischen Missense-Varianten (32). Funktionell wurde die Pathogenität der identifizierten Varianten in vitro nachgewiesen, was zu intrazellulärem Mis-Trafficking und beeinträchtigter Transportaktivität führte (32). Defektes SLC26A1 stellt daher eine neue Ursache für CaOx-NL dar und sollte bei der Untersuchung von Personen auf Ursachen für wiederkehrende CaOx-Steinbildung in Betracht gezogen werden.
Neues klinisches Patientenregister für erbliche Nierensteinerkrankungen
Die meisten epidemiologischen Daten über die zunehmende Prävalenz in westlichen Ländern stammen aus US-amerikanischen Datenbanken. Obwohl sie dringend benötigt werden, sind zentralisierte europäische Datenbanken derzeit nicht verfügbar. Da die oben erwähnten genetischen Studien zur Prävalenz der erblichen Nierensteinerkrankung mit kleinen Kohorten aus spezialisierten Zentren sowohl in Europa als auch in den USA durchgeführt wurden, ist eine Übertragung auf die allgemeine Situation in Europa nicht zulässig. Während in den USA das Rare Kidney Stone Consortium eine Plattform darstellt, die Register-, Grundlagenforschungs- und klinische Forschungsaktivitäten für seltene Erkrankungen wie Cystinurie, PH, APRT-Mangel, Dent- und Lowe-Krankheit integriert und koordiniert, gibt es bis heute weder in Europa noch in Deutschland eine vergleichbare Datenerhebung über Patienten mit erblichen Nierensteinerkrankungen. In Zusammenarbeit mit dem bestehenden europäischen PH-Register OxalEurope (Prof. Bernd Hoppe, Universität Bonn) und mit finanzieller Unterstützung der Deutschen Forschungsgemeinschaft und der Else Kröner-Fresenius Stiftung haben wir vor kurzem ein klinisches Patientenregister für erbliche Nierensteinerkrankungen“ an der Universität Leipzig eingerichtet. Das Register wird bundesweit von der Deutschen Gesellschaft für Nephrologie des Erwachsenenalters (DGfN) und der Pädiatrischen Nephrologie (GPN) unterstützt. Es ist außerdem im Deutschen Register für Klinische Studien (DRKS-ID: DRKS00012891) registriert. Als grundlegender Teil der Studienrekrutierung wird eine Hochdurchsatz-Mutationsanalyse für bekannte und neue Nierensteingene auf Forschungsbasis für Patienten ohne etablierte molekulare Diagnose, aber mit einem klinischen Bild, das auf eine zugrundeliegende genetische Anfälligkeit hinweist, angeboten: z.B. frühes Erkrankungsalter (<40 Jahre), positive Familienanamnese, indikative Phänotypen wie NC, Cystinurie oder RTA und schwer rezidivierende NL (>3×) (Tabelle 2). Während Patienten mit einer bereits gestellten genetischen Diagnose in der Regel aufgenommen werden, werden Fälle mit sekundären NL/NC-Ursachen wie Malignität, Sarkoidose und primärer Hyperparathyreoidismus nicht in die genetische Analyse einbezogen. Für die aktive Aufnahme von Patienten benötigt ein klinisches Zentrum in der Regel die Genehmigung des lokalen Institutional Review Board; ein Verfahren, für das wir unsere Hilfe und Unterstützung anbieten, indem wir entsprechende Vorlagen bereitstellen. Nach der ethischen Genehmigung können die Einverständniserklärung und die klinischen Datenblätter (z. B. der klinische Fragebogen) von unserer Register-Website (http://www.mks-registry.net) heruntergeladen werden. Um eine gründliche klinische Phänotypisierung zu gewährleisten, fragen wir unter anderem nach ethnischer Zugehörigkeit, Blutsverwandtschaft, Familienanamnese, Alter des Auftretens, Rezidiv (definiert als jedes vermeintlich neue Nierensteinereignis), täglicher Flüssigkeitsaufnahme, chirurgischen Eingriffen und extrarenaler Beteiligung. Die Dokumente können vom Patienten mit Hilfe des behandelnden Arztes ausgefüllt werden. Darüber hinaus werden biochemische Serumparameter, einschließlich Kreatinin, eGFR, PTH, Vitamin D, Elektrolyte, Harnsäure und Urinanalyse (pH-Wert, Kalzium, Phosphat, Magnesium, Harnsäure, Citrat, Oxalat und Cystin, vorzugsweise aus 24-Stunden-Urin, wenn nicht aus Punkturin), sowie Daten zur Analyse der Steinzusammensetzung bei der Einschreibung angefordert. Da eine 24-Stunden-Urinsammlung und eine Analyse der Steinzusammensetzung nicht in allen Einrichtungen routinemäßig durchgeführt wird, beziehen wir diese Parameter bei Verfügbarkeit mit ein. Wünschenswert, aber nicht verpflichtend, sind 2-jährliche klinische Nachuntersuchungen der eingeschriebenen Patienten. Nach der Registrierung erhalten die einladenden klinischen Zentren ein persönliches Login, um die Patientendaten über unsere Register-Website (http://www.mks-registry.net) einzugeben. Alternativ bieten wir an, die Daten elektronisch zu erfassen, wenn sie uns in Papierform zugesandt werden. Die eingegebenen Daten werden auf einem gesicherten Server gespeichert und können von den teilnehmenden klinischen Zentren abgerufen werden, um ihre eigenen Patientendaten einzusehen. Die folgenden Websites bieten weitere Informationen:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
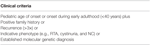
Table 2. Einschlusskriterien für die Mutationsanalyse im klinischen Patientenregister.
Zusammenfassend lässt sich sagen, dass die Nierensteinerkrankung eine zunehmend verbreitete Erkrankung ist, die klinisch heterogen ist und deren genetische Ursachen nur unzureichend verstanden werden. Wie eine Reihe neuerer Studien gezeigt hat, wird die Prävalenz monogener Erkrankungen höchstwahrscheinlich unterschätzt. Durch die Einrichtung eines zentralen Patientenregisters für erbliche Nierensteinleiden werden wir dazu beitragen, die große Wissenslücke über die Genetik von Nierensteinleiden zumindest teilweise zu schließen. In diesem Zusammenhang sind klinische Register aus mehreren Gründen wertvolle Quellen: Erstens wird die Beschreibung besserer Phänotyp-Genotyp-Assoziationen für eine präzisere Patientenstratifizierung in zukünftigen klinischen Forschungsstudien entscheidend sein. Zweitens wird die Identifizierung neuer Krankheitsgene mit neuen Krankheitsmechanismen die Lücke der unbekannten NL/NC-Ätiologie verkleinern; und drittens trägt die Entschlüsselung neuer molekularer Ziele dazu bei, den Weg für die Entwicklung von Medikamenten zur Rezidivprävention in schwer betroffenen Familien zu ebnen.
Ethikerklärung
Diese Studie wurde in Übereinstimmung mit den Empfehlungen der „Ethikkommission an der Medizinischen Fakultät der Universität Leipzig“ mit schriftlicher Einwilligung aller Probanden durchgeführt. Alle Probanden gaben ihre schriftliche Einwilligung nach Aufklärung gemäß der Deklaration von Helsinki. Das Protokoll wurde von der „Ethikkommission an der Medizinischen Fakultät der Universität Leipzig“ genehmigt.
Beiträge der Autoren
JH hat das Manuskript konzipiert und geschrieben. BH, AS, LM und RS redigierten das Manuskript und bauten die Infrastruktur des Registers auf, die dem Leser vorgestellt wird.
Erklärung zu Interessenkonflikten
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Finanzierung
Die Forschung wurde durch Projektförderungen der DFG (HA 6908/2-1) und des EKFS (2016_A52) an JH finanziert. Diese Arbeit wurde außerdem durch das Bundesministerium für Bildung und Forschung (BMBF), Deutschland, FKZ: 01EO1501 an JH unterstützt.
1. Scales CD Jr., Smith AC, Hanley JM, Saigal CS. Prävalenz von Nierensteinen in den Vereinigten Staaten. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Genetik der hypercalciurischen Nephrolithiasis: Nierensteinerkrankung. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Progress in understanding the genetics of calcium-containing nephrolithiasis. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sequenzvarianten im CLDN14-Gen assoziieren mit Nierensteinen und Knochenmineraldichte. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 beeinflusst die Harnsäurekonzentration mit ausgeprägten geschlechtsspezifischen Effekten. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutationen im Glukose-Transporter 9-Gen SLC2A9 verursachen renale Hypourikämie. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identifizierung und Charakterisierung eines Gens mit Basensubstitutionen, die mit dem Phänotyp der absorptiven Hyperkalziurie und einer niedrigen Knochendichte der Wirbelsäule in Verbindung stehen. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Falsches Targeting der peroxisomalen L-Alanin:Glyoxylat-Aminotransferase in Mitochondrien bei Patienten mit primärer Hyperoxalurie hängt von der Aktivierung einer kryptischen mitochondrialen Targeting-Sequenz durch eine Punktmutation ab. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Human adenine phosphoribosyltransferase. Identifizierung von allelischen Mutationen auf Nukleotidebene als Ursache für einen vollständigen Mangel des Enzyms. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutationen in ATP6N1B, die für eine neue vakuoläre 116-kD-Untereinheit der Nierenprotonenpumpe kodieren, verursachen rezessive distale renale tubuläre Azidose mit erhaltenem Gehör. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutationen in dem Gen, das für die B1-Untereinheit der H+-ATPase kodiert, verursachen eine renale tubuläre Azidose mit sensorineuraler Taubheit. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II deficiency syndrome in a Belgian family is caused by a point mutation at an invariant histidine residue (107 His→Tyr): complete structure of the normal human CA II gene. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. A common molecular basis for three inherited kidney stone diseases. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutationen im Chloridkanal-Gen, CLCNKB, verursachen das Bartter-Syndrom Typ III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutationen im Tight-Junction-Gen Claudin 19 (CLDN19) sind mit renalem Magnesiumverlust, Nierenversagen und schwerer Augenbeteiligung assoziiert. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutationen in CYP24A1 und idiopathische infantile Hyperkalzämie. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Das Gen, das für Hydroxypyruvat-Reduktase (GRHPR) kodiert, ist bei Patienten mit primärer Hyperoxalurie Typ II mutiert. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a β cell phenotype. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutationen in DHDPSL sind verantwortlich für primäre Hyperoxalurie Typ III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identification of 17 independent mutations responsible for human hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Tightly linked flanking markers for the Lowe oculocerebrorenal syndrome, with application to carrier assessment. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Das Bartter-Syndrom, hypokaliämische Alkalose mit Hyperkalziurie, wird durch Mutationen im Na-K-2Cl-Cotransporter NKCC2 verursacht. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutationen in SLC26A1 verursachen Nephrolithiasis. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis und Osteoporose im Zusammenhang mit Hypophosphatämie, verursacht durch Mutationen im Natrium-Phosphat-Cotransporter Typ 2a. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Non-Type I Cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identifizierung von zwei Mutationen im menschlichen Xanthin-Dehydrogenase-Gen, die für die klassische Xanthinurie Typ I verantwortlich sind. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract |Ref Full Text | Google Scholar
42. Hildebrandt F. Genetic kidney diseases. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutationen in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Klinische und biochemische Phänotypen von Erwachsenen mit monoallelischen und biallelischen CYP24A1-Mutationen: Nachweis eines Gendosiseffekts. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Neueste diagnostische und therapeutische Erkenntnisse. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolithiasis und Hepatotoxizität sind mit dem Anionentransporter Sat1 in Mäusen verbunden. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | Querverweis Volltext | Google Scholar