John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1Escuela de Ciencias Biomédicas, Kent State University, Ohio, USA
2Departamento de Ciencias Biológicas, Kent State University, Ohio, USA
Abstracto
Cada vez hay más pruebas que resaltan la íntima relación entre la diabetes tipo II (T2D) y la enfermedad de Alzheimer (AD). Es importante destacar que estas dos enfermedades comparten una serie de similitudes patológicas, incluyendo la acumulación de amiloide, el estrés oxidativo, la inflamación y la muerte celular. Hasta la fecha, no existen terapias farmacológicas para la EA y la T2D y existe una necesidad crucial de descubrir y desarrollar nuevas terapias para estas enfermedades. Varios estudios en humanos y en roedores han demostrado que la suplementación con hormonas metabólicas es muy valiosa para mejorar la función cognitiva y la salud metabólica general tanto en la T2D como en la EA. La hormona pancreática amilina ha surgido como un componente crucial de la etiología de la enfermedad tanto de la T2D como de la EA, aunque el papel exacto que desempeña la amilina en estas enfermedades aún no se conoce bien. Aquí, revisamos críticamente la literatura actual que utiliza la amilina humana o su análogo sintético, pramlintida, así como los antagonistas del receptor de amilina para el tratamiento de la EA.
Introducción
La enfermedad de Alzheimer (EA) es una enfermedad neurodegenerativa progresiva y debilitante que se caracteriza por la acumulación de placas de beta-amiloide (Aβ) y ovillos neurofibrilares compuestos por tau1 hiperfosforilada. La acumulación de estos péptidos patológicos contribuye a los déficits de las funciones ejecutivas, como el aprendizaje y la memoria, el estado de ánimo, el afecto, etc., y supone una carga sustancial para el paciente y los cuidadores. La incidencia de la EA está aumentando a un ritmo alarmante en los Estados Unidos, con una estimación de 5,5 millones de estadounidenses que viven con EA en 2017 y se espera que esta cifra se triplique para el año 20502. Además, el coste de la atención y el tratamiento de los pacientes con EA supera actualmente los 200.000 millones de dólares anuales y se espera que aumente3. Aunque la EA es claramente un problema monumental dentro y fuera de los Estados Unidos, las opciones de tratamiento siguen siendo muy limitadas4. Se han llevado a cabo muchos ensayos de fármacos con una amplia gama de enfoques dirigidos, pero actualmente sólo hay seis fármacos aprobados por la FDA para la EA y son únicamente tratamientos sintomáticos5, 6. Hasta la fecha, la mayoría de los agentes farmacológicos desarrollados se han dirigido específicamente a la patología distintiva Aβ o tau, pero ninguno ha tenido éxito en la eliminación o prevención de la patología4. Por ello, existe una necesidad fundamental de desarrollar tratamientos terapéuticos y preventivos viables para la EA.
La EA relacionada con la edad (esporádica) es una enfermedad multifactorial complicada, que tiene numerosas influencias genéticas y ambientales. El entorno y el estilo de vida están fuertemente implicados en el desarrollo de la EA esporádica; factores como la dieta7-9, la obesidad8-10, el síndrome metabólico7, la diabetes de tipo II (T2D)9, 11 y las enfermedades cardiovasculares12 han sido implicados en la causalidad de la EA. Es fundamental que las tasas de obesidad y diabetes aumenten rápidamente en paralelo a la EA12, 13. Aunque la relación entre la obesidad y la EA no está muy clara, hay pruebas de que la obesidad en la mediana edad desempeña un papel en el desarrollo de la EA10. Y lo que es más importante, la obesidad suele ir acompañada de otras enfermedades, como las cardiovasculares, la hipertensión, la dislipidemia, la T2D, el ictus, etc.14. La incidencia de la T2D está aumentando rápidamente, ya que los CDC estiman que aproximadamente 30,3 millones de personas (1 de cada 10 adultos) en los EE.UU. tienen diabetes y la asombrosa cifra de 84,1 millones (1 de cada 3 adultos) tienen prediabetes, la mayoría de los cuales no son conscientes de su condición. Además, debido a la disminución a gran escala de la actividad física, acompañada de un aumento simultáneo de la ingesta de alimentos y de una dieta inadecuada, se prevé que las tasas de obesidad, T2D, síndrome metabólico y enfermedades cardiovasculares aumenten hasta alcanzar una cifra estimada de 600 millones de casos de T2D en todo el mundo para el año 203515.
El conjunto de pruebas que implican la función y la enfermedad metabólicas en el proceso de deterioro cognitivo y envejecimiento es considerable16, 17. Por ejemplo, aproximadamente el 70% de las personas diagnosticadas de T2D presentan un deterioro cognitivo y un número considerable de pacientes con T2D desarrollan posteriormente demencia16, 18-21. Los individuos diagnosticados de T2D durante al menos cinco años tienen un riesgo significativamente mayor de desarrollar EA en comparación con aquellos que han padecido T2D durante menos de cinco años17. En conjunto, estos datos sugieren que la creciente prevalencia de la T2D en la población puede estar contribuyendo al aumento de las tasas de EA.
La T2D se caracteriza inicialmente por un nivel elevado de glucosa e insulina en la sangre, lo que conduce a una hiperinsulinemia; es importante destacar que la amilina, una pequeña hormona metabólica producida por las células de los islotes β del páncreas, está empaquetada y segregada conjuntamente con la insulina, por lo que se produce en exceso en la T2D22. Es importante destacar que hay una serie de características patológicas que están presentes tanto en la T2D como en la EA: 1) disminución del metabolismo cerebral y resistencia hormonal metabólica 2) patología amiloide 3) estrés oxidativo (SO) e inflamación. La hiperinsulinemia e hiperamilinemia crónicas conducen a una serie de problemas fisiológicos: la hiperinsulinemia crónica conduce a la resistencia a la insulina del sistema22, a la alteración del transporte de insulina a través de la barrera hematoencefálica (BBB)23, 24 y, por tanto, a la disminución de la señalización de la insulina dentro del cerebro25. La pérdida de la señalización de la insulina en el cerebro se asocia con una serie de características patológicas relacionadas con la EA, incluyendo el aumento de la producción de Aβ, la fosforilación de tau y la neuroinflamación.
Además, la amilina comparte características patológicas similares con el Aβ en altas concentraciones26 y puede ser una vía común entre las dos enfermedades. Por ejemplo, se han encontrado fibrillas de amilina en el páncreas del 95% de los pacientes con T2D27-29 y causan una serie de alteraciones fisiológicas que incluyen la afluencia aberrante de Ca2+, el aumento de la secreción de citoquinas proinflamatorias30,31 y, en última instancia, la pérdida de células de los islotes β32. Además, la amilina atraviesa fácilmente la BBB y forma fibrillas de amilina, así como placas mixtas con Aβ dentro del cerebro, y puede ser responsable de la patología similar a la EA y de la siembra de Aβ en la T2D33-35. Se sabe que la amilina afecta a la potenciación a largo plazo (LTP) en el hipocampo y puede tener una influencia innata en la función cognitiva dentro del cerebro36-39. Sin embargo, sigue sin estar claro si la amilina es un insulto tóxico en estas enfermedades o si su pérdida funcional a través de la agregación o la pérdida de células β en la fase tardía de la T2D contribuye al desarrollo de una EA.
La dicotomía de la señalización de la amilina
Aún hay mucho debate sobre la implicación del receptor de la amilina (AMYR) y la señalización de la amilina en la progresión de la enfermedad y la etiología de la T2D y la EA. El conjunto de investigaciones destinadas a discernir esta relación se está ampliando rápidamente. Todas las investigaciones pertinentes han demostrado sistemáticamente que la modulación de la señalización de la amilina afecta a la patología relacionada con la EA. Sin embargo, la naturaleza de esta relación aún no se ha dilucidado de forma concreta. Varios grupos han aportado datos convincentes que sugieren que la señalización de la amilina es beneficiosa para prevenir la patología relacionada con la EA y los déficits cognitivos tanto in vivo como in vitro40-44. Es importante destacar que la pramlintida, una forma recombinante no agregada de amilina, que se utiliza junto con las terapias de insulina para tratar la diabetes y mejora el control glucémico, reduce el peso corporal y disminuye los marcadores séricos de la OS45-47, también resulta prometedora como terapia para la EA. Sin embargo, hasta la fecha no ha habido ningún ensayo clínico que haya pretendido utilizar la amilina o la pramlintida como agente terapéutico en el tratamiento de la demencia. Las evidencias claras de los estudios con roedores sugieren que el tratamiento crónico con amilina o pramlintida humana supone un fuerte beneficio terapéutico en la reducción de la patología relacionada con la EA; la suplementación con amilina/pramlintida reduce los niveles de Aβ soluble, la carga de placa, la fosforilación de tau, la neuroinflamación y la OS, al tiempo que mejora la cognición40-42,44. Los datos anteriores sugieren que una pérdida de la señalización innata de la amilina en el SNC debido a la agregación da lugar a un mayor riesgo de desarrollo de la EA y se trata con más detalle en Grizzanti et al. 201848.
En cambio, los estudios también muestran que la amilina humana y el Aβ tienen efectos tóxicos similares y que estos efectos tóxicos pueden aliviarse utilizando el antagonista AMYR36-39,49. Por ejemplo, los datos muestran que el tratamiento in vivo con antagonistas de AMYR produce beneficios fisiológicos muy similares al tratamiento con amilina o pramlintida. El tratamiento de ratones con EA TgCRND8 con AC253, un antagonista de AMYR, o su homólogo cAC253 cíclico, reduce la neuroinflamación, los niveles de Aβ soluble y la carga de placas, al tiempo que mejora la cognición50. Del mismo modo, los estudios in vitro/ex-vivo muestran que dosis bajas de amilina humana o Aβ causan interrupciones en la LTP y que estos déficits son bloqueados por AC253 o pramlintida38,39, y dosis más altas de amilina humana/oligómeros de amilina se asocian con la afluencia incontrolada de Ca2+, que está fuertemente relacionada con la muerte celular26,32. En conjunto, estos datos apoyan una función tóxica de los oligómeros de amilina y, por lo tanto, un potencial mecanismo terapéutico para el bloqueo del AMYR. Por el contrario, otros han demostrado que los efectos beneficiosos de la amilina pueden bloquearse con AC25341. Así pues, el potencial terapéutico del tratamiento o la inhibición de la amilina sigue sin estar claro y pone de manifiesto la naturaleza compleja y dicotómica de los amiloides en el cerebro y la periferia.
Reconstruyendo el rompecabezas
Hay una serie de agujeros en la bibliografía actual que es necesario llenar para dar una imagen más completa de la historia de la amilina: 1) la naturaleza del sistema innato de amilina y la señalización de amilina dentro del cerebro 2) las capacidades de señalización de Aβ y pramlintida a través de los tres principales receptores AMYR y relacionados 3) los mecanismos terapéuticos por los que median la inhibición de amilina/pramlintida o AMYR. En primer lugar, nuevos e interesantes datos demuestran que el AMYR no sólo participa en la señalización, sino también en el transporte del ligando a través de la BBB. El AMYR es un receptor heterodimérico compuesto por un receptor de calcitonina y una proteína modificadora de la actividad del receptor (1-3)51. En este sentido, un knockdown global del 50% del receptor de calcitonina (un componente clave del AMYR) redujo significativamente la cantidad de AC253 encontrada en el cerebro50, lo que indica que los AMYR localizados en la BBB están implicados en el transporte de estos ligandos al cerebro y también pueden estar implicados en el traslado de amilina y pramlintida dentro/fuera del cerebro. La existencia de estos mecanismos de transporte en la BBB sugiere que la amilina tiene probablemente una función fisiológica innata en el cerebro, ya que su transporte al cerebro está estrechamente controlado. Sin embargo, todavía no está claro cómo la señalización de la amilina o la falta de ella conduce a las características patológicas de la EA y si el AMYR es el vehículo a través del cual Aβ media sus efectos tóxicos.
A continuación, existen pruebas contradictorias con respecto a la relación entre Aβ y el AMYR. Aunque varios estudios demuestran claramente que la amilina humana y el Aβ tienen efectos similares sobre la LTP en el SNC y el uso de inhibidores del AMYR mejora estos efectos deletéreos36-39, otras evidencias sugieren que el Aβ (1-42) es incapaz de señalizar a través de cualquiera de los AMYR para evocar cualquier tipo de respuesta de AMPc en una amplia variedad de concentraciones52. Es posible que Aβ active diferentes cascadas de señalización a través de la interacción con el AMYR o que simplemente actúe como un inhibidor competitivo inerte, pero esto aún está por demostrar.
Además, un estudio separado demostró que la amilina oligomérica media sus efectos tóxicos directamente a través del AMYR e indirectamente a través del TRPV4, un canal de cationes no selectivo26. Concentraciones bajas de amilina humana evocan una respuesta de Ca2+ que está mediada por su receptor nativo. Sin embargo, a concentraciones más altas, la amilina humana forma oligómeros y activa una señalización aberrante que da lugar a la activación de los canales TRVP4 y permite la afluencia incontrolada de cationes, en particular de Ca2+. El bloqueo farmacológico del AMYR y del TRPV4 demuestra que ambos receptores son necesarios para que la amilina humana oligomérica induzca sus efectos tóxicos sobre el Ca2+26. Como tal, es probable que Aβ medie sus efectos tóxicos sobre el AMYR de manera similar, aunque estos datos aún no existen. La afluencia incontrolada de Ca2+ está relacionada con una serie de fenómenos patológicos, como la liberación vesicular incontrolada, la disfunción del sistema operativo y mitocondrial, la apoptosis, etc. En este sentido, es probable que la disfunción celular y el desarrollo de patología adicional similar a la EA que surge de la señalización tóxica del amiloide estén mediados tanto por el AMYR como por el TRPV4. Por ello, es necesario discernir las cascadas de señalización que modulan la relación entre el AMYR y el TRVP4. Además, están garantizados los experimentos farmacológicos que utilizan Aβ y pramlintida en una amplia gama de dosis para determinar los efectos de Aβ y pramlintida en las corrientes de Ca2+, LTP, producción de AMPc y otras cascadas de señalización para determinar sus capacidades de señalización. Estos experimentos ayudarán a llenar algunos de los vacíos en la literatura actual con respecto al AMYR y su participación en los estados de enfermedad (Figura 1).
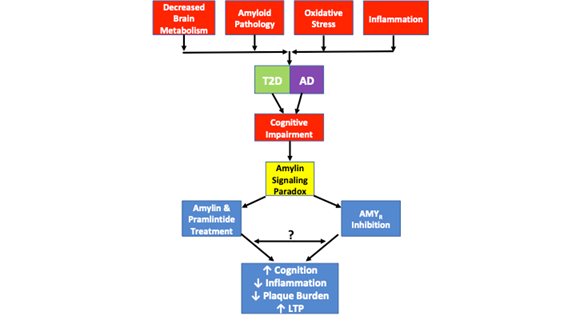
La Figura 1. representa la paradoja de señalización de la amilina y las similitudes patológicas observadas en la T2D y la EA. La disminución del metabolismo cerebral, la patología amiloide, el estrés oxidativo y la inflamación son características patológicas comunes observadas en ambas enfermedades. Aunque no todos los casos de T2D o EA incluyen cada una de estas características patológicas, todos los casos presentan un deterioro cognitivo. La paradoja de la señalización de la amilina entra en juego, ya que los estudios han demostrado que tanto la inhibición de la AMYR como el agonismo de la AMYR mediante el tratamiento con amilina y pramlintida dan lugar a una mejora de la cognición, una disminución de la inflamación, una reducción de la carga de placa y un aumento de la LTP. Los mecanismos de señalización gobernados por el agonismo de AMYR y el antagonismo de AMYR aún no han sido completamente dilucidados. Mientras que la señalización de la amilina se asocia tradicionalmente con el AMPc y la señalización de la PKA, no está claro si otras cascadas también son activadas por la amilina/pramlintida. Además, no está claro si los antagonistas del AMYR, los oligómeros de amilina o el Aβ señalan a través del AMYR o si hay alguna similitud o diafonía entre todos estos ligandos del AMYR. Como tal, una serie de experimentos propuestos en esta revisión ayudarán a dilucidar aún más la verdadera naturaleza del AMYR.
Conclusiones
La disparidad actual con respecto al papel de la señalización de la amilina en el cerebro demuestra una necesidad esencial de dilucidar aún más la participación de la amilina tanto en la EA como en la T2D. En la T2D, es probable que en las primeras fases de la enfermedad, la amilina inunde el cerebro, forme oligómeros, induzca una señalización aberrante a través de su receptor nativo y reclute al TRPV4 para inducir una afluencia patológica de Ca2+ que dé lugar a una disfunción neuronal generalizada que se manifieste como OS, liberación vesicular incontrolada y disfunción interneuronal, inflamación y la consiguiente muerte celular. Este mecanismo puede ser responsable de la transición inicial del cerebro sano al envejecimiento cerebral en la enfermedad metabólica. Por ello, la inhibición del AMYR o del TRVP4 en determinados momentos de la enfermedad metabólica y en las primeras fases de la diabetes puede estar justificada para bloquear los efectos tóxicos de la amilina o del Aβ oligomérico. Sin embargo, también hay pruebas sólidas que sugieren que la sustitución de la amilina por amilina humana o pramlintida reduce la mayor parte de la patología principal relacionada con la EA, al tiempo que mejora la cognición en modelos de roedores de la EA. Por lo tanto, la sustitución de la señalización de la amilina con amilina o pramlintida en las etapas medias y tardías de la diabetes, cuando la señalización de la amilina se pierde debido a la agregación, la oligomerización o la pérdida de células β, puede estar justificada. Para ello, también es necesario discernir la presentación temporal de los eventos patológicos en el envejecimiento cerebral metabólicamente vinculado y las opciones terapéuticas para la enfermedad en etapas tempranas, intermedias y tardías. El análisis crítico y la comprobación de la naturaleza directa y las capacidades de señalización de estos amiloides, así como la naturaleza terapéutica de los tratamientos temporales específicos, pueden ayudar a salvar la distancia entre las terapias de inhibición de AMYR y las terapias de sustitución de amilina.
Financiación
La financiación de este artículo fue proporcionada por la subvención 1R15AG050292-01A1 de los Institutos Nacionales del Envejecimiento.
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau y disfunción sináptica. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013; 80: 1778-1783.
- Asociación As. 2016 Datos y cifras de la enfermedad de Alzheimer. Alzheimer’s & Dementia. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Desarrollo de fármacos para la enfermedad de Alzheimer: pocos candidatos, fracasos frecuentes. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolución de la evidencia sobre la eficacia y la rentabilidad de los inhibidores de la acetilcolinesterasa y la memantina para la enfermedad de Alzheimer: revisión sistemática y modelo económico. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezilo y memantina para la enfermedad de Alzheimer de moderada a grave. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Niveles de glucosa y riesgo de demencia. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Obesidad en la mediana edad y demencia: metaanálisis y previsión ajustada de la prevalencia de la demencia en Estados Unidos y China. Obesity. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Diario de investigación de la diabetes. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesidad en la mediana edad y riesgo futuro de demencia: un estudio longitudinal de 27 años basado en la población. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. La diabetes tipo 2 como factor de riesgo para la enfermedad de Alzheimer: los factores de confusión, las interacciones y la neuropatología asociada a esta relación. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. La exposición a factores de riesgo vascular en la mediana edad acelera el envejecimiento estructural del cerebro y el deterioro cognitivo. Neurology. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus En Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Obesidad y sus condiciones comórbidas. Clin Cornerstone. 1999; 2: 17-31.
- Federación ID. Atlas de la diabetes de la FID. Bruselas: Federación Internacional de Diabetes. 2013.
- K Dash S. Cognitive impairment and diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Demencia y declive cognitivo en la diabetes tipo 2 y en los estadios prediabéticos: hacia intervenciones dirigidas. La lanceta Diabetes & endocrinología. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus y enfermedad de Alzheimer: patología y tratamiento compartidos. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalencia de la diabetes y la prediabetes y sus factores de riesgo entre los adultos de Bangladesh: una encuesta nacional. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Actas de la Academia Nacional de Ciencias. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer’s disease. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Objetivo múltiple de hAmylin en las neuronas primarias del hipocampo de rata. Neurofarmacología. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mecanismos que relacionan la obesidad con la resistencia a la insulina y la diabetes tipo 2. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. La revista americana de patología. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Marcadores inflamatorios y riesgo de diabetes tipo 2: una revisión sistemática y meta-análisis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. El β-amiloide de Alzheimer, la amilina de los islotes humanos y el fragmento de proteína priónica evocan elevaciones de calcio libre intracelular por un mecanismo común en una línea celular neuronal de GnRH hipotalámica. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Díaz E, et al. Depósito de amilina en el cerebro: un segundo amiloide en la enfermedad de Alzheimer. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. Siembra in vivo y siembra cruzada de amiloidosis localizada: un vínculo molecular entre la diabetes tipo 2 y la enfermedad de Alzheimer. La revista americana de patología. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Receptor de amilina: una diana fisiopatológica común en la enfermedad de Alzheimer y la diabetes mellitus. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. El péptido amiloide β (Aβ) activa directamente el subtipo de receptor de amilina-3 desencadenando múltiples vías de señalización intracelular. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide antagoniza la depresión de la potenciación a largo plazo del hipocampo inducida por el beta amiloide (Aβ) y la amilina humana. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. La depresión inducida por el beta amiloide de la potenciación a largo plazo del hipocampo está mediada a través del receptor de amilina. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. La inyección intraperitoneal del péptido pancreático amilina reduce potentemente el deterioro del comportamiento y la patología amiloidea cerebral en modelos murinos de la enfermedad de Alzheimer. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Los ligandos del receptor de amilina reducen la cascada patológica de la enfermedad de Alzheimer. Neurofarmacología. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Efectos neuroprotectores del análogo de la amilina pramlintida en la patogénesis de la enfermedad de Alzheimer y la cognición. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. La desestabilización de β-catenina por mutaciones en presenilina-1 potencia la apoptosis neuronal. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Acetato de pramlintida inyectable para el tratamiento de la diabetes mellitus tipo 1 y tipo 2. Clin Ther. 2007; 29: 535-562.
- Singh-Franco D, Pérez A, Harrington C. El efecto del acetato de pramlintida sobre el control glucémico y el peso en pacientes con diabetes mellitus tipo 2 y en pacientes obesos sin diabetes: una revisión sistemática y metaanálisis. Diabetes, Obesidad y Metabolismo. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide como complemento del tratamiento con insulina mejora el control glucémico y del peso a largo plazo en pacientes con diabetes tipo 2. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Acciones de la proteína β-amiloide en las neuronas humanas se expresan a través del receptor de amilina. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. El AC253 cíclico, un nuevo antagonista del receptor de amilina, mejora los déficits cognitivos en un modelo de ratón de la enfermedad de Alzheimer. Alzheimer’s & Dementia: Investigación Traslacional & Intervenciones Clínicas. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reducción del comportamiento nociceptivo en ratones knockout del polipéptido amiloide de los islotes (amilina). Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Activity of pramlintide, rat and human amylin but not Aβ1-42 at human amylin receptors. Endocrinology. 2014; 155: 21-26.