Introducción
Las mutaciones de novo en la línea germinal (DNMs) son cambios genéticos en el individuo causados por la mutagénesis que ocurre en los gametos parentales durante la oogénesis y la espermatogénesis. Aquí, el término «de novo» no debe confundirse con el término «mutación nueva». A pesar de que las DNM en el contexto de un trío (padre, madre e hijo) son mutaciones novedosas, pueden ser variantes comunes, raras o nuevas en la población general. Para medir y explicar la tasa de un DNM concreto, es necesario evaluar primero el impacto en el fenotipo de la variante, porque pueden evolucionar nuevos rasgos favorables cuando las mutaciones genéticas que surgen ofrecen un beneficio específico para la supervivencia (Front Line Genomics, 2017).
En los seres humanos con enfermedades genéticas no mendelianas que se producen de forma esporádica, los DNM suelen ser novedosos, más fiables y más perjudiciales que las variantes heredadas porque no están sometidos a una fuerte selección natural (Crow, 2000; Front Line Genomics, 2017). Por lo tanto, identificar la causa genética de un trastorno inducido por una DNM en un individuo puede ser un reto desde el punto de vista clínico, porque la pleiotropía y la heterogeneidad genética pueden subyacer a un único fenotipo (Eyre-Walker y Keightley, 2007). En consecuencia, en la última década se han realizado esfuerzos considerables para secuenciar exomas de individuos con enfermedades de etiología genética poco clara con el fin de realizar diagnósticos clínicos. Sin embargo, incluso tras la detección de variantes de novo candidatas, sigue habiendo información insuficiente sobre las variantes comunes y raras, lo que impide llegar a una conclusión clara sobre la patogenicidad de la variante de novo identificada y su papel en la enfermedad (Acuna-Hidalgo et al., 2016). Esta limitación puede explicarse por el hecho de que las variantes de novo suelen ser heterocigotas y pueden ser extremadamente raras o comunes. En los casos de variantes de novo muy raras, la patogenicidad de la variante puede ser difícil de probar ya que no hay más pacientes con el mismo fenotipo y variante de novo. En los casos de variantes de novo comunes, es posible que no se conozcan los factores que determinan las manifestaciones de la patogenicidad de la variante, sobre todo si algunos individuos de la población general tienen la variante pero no presentan la enfermedad genética. Sin embargo, independientemente de la tasa de variantes de novo, ambos tipos de variantes pueden escalarse sobre la base de la aptitud relativa y la selección natural.
La adaptación depende de muchos factores; por lo tanto, para evaluar si una DNM es patógena o adaptativa, y para entender por qué se produce con una frecuencia particular en la población, es necesario examinar la variante en condiciones adecuadas. Estas incluyen el entorno, la edad de los padres, el contexto genómico, la epigenética y otros factores, ya que todos ellos influyen en el valor de la aptitud relativa media que aumenta monotónicamente, mientras que la fuerza de la selección disminuye (Peck y Waxman, 2018).
El objetivo principal de este estudio fue dilucidar la tasa de ocurrencia de las DNM y determinar cómo se distribuyen estas mutaciones en los exomas de la población lituana general. También examinamos si la frecuencia de estas mutaciones se veía afectada por la composición o los parámetros estructurales de las secuencias en las que se producían y por otros factores que pudieran influir en los mecanismos subyacentes a la formación de estos DNM. Por último, tratamos de establecer si los DNMs surgieron debido a la intensa presión de la selección natural sobre las regiones funcionales. Aunque la distribución y la intensidad de los DNMs han sido objeto de muchos estudios, no se habían explorado previamente en la población lituana.
Materiales y métodos
En este estudio, analizamos muestras de la población lituana obtenidas del proyecto LITGEN (LITGEN, 2011). El conjunto de datos consistió en 49 tríos con un total de 144 individuos diferentes. El ADN genómico se extrajo de la sangre venosa utilizando el método de extracción con fenol-cloroformo o la plataforma de extracción de ADN automatizada TECAN Freedom EVO® (Tecan Schweiz AG, Suiza) basada en el método de partículas paramagnéticas. Los exomas se secuenciaron en un sistema de secuenciación SOLiD 5500 (lecturas de 75 pb). Los datos de secuenciación se procesaron y prepararon con el software Lifescope. Los exomas se mapearon de acuerdo con el genoma humano de referencia 19. La profundidad media de las lecturas de secuenciación fue de 38,5. Los archivos con formato BAM de la madre, el padre y el hijo generados por Lifescope se combinaron utilizando el software SAMtools para cada trío.
Las mutaciones de novo se identificaron mediante dos programas de software: VarScan (Koboldt et al., 2012) y VarSeqTM. Se consideró que una variante potencial era una DNM si se identificaba en la descendencia pero no estaba presente en ninguno de los padres en la misma posición. En total, VarScan y VarSeqTM detectaron 1.752 y 4.756 DNM, respectivamente. Para descartar las llamadas de novo falsas positivas, cuando no se sabía si todos los individuos del trío se habían identificado correctamente, se aplicaron filtros conservadores sobre los parámetros de calidad de los DNM detectados, como sigue (1) calidad del genotipo del individuo ≥50; (2) número de lecturas en cada sitio >20. Se utilizó el software SnpSift para aplicar estos filtros a los datos generados por VarScan. Los datos generados por el software VarSeqTM se filtraron eligiendo los mismos parámetros de filtrado en el segmento Trio Workflow. Además, para descartar las variantes restantes que eran somáticas (solo presentes en una fracción de las células sanguíneas secuenciadas) con bajo balance alélico o artefactos de secuenciación, los DNM se filtraron estableciendo un umbral para la fracción observada de las lecturas en individuos con el alelo alternativo (el balance alélico) para el trío (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Además, todas las posibles variantes de nucleótido único identificadas y filtradas fueron revisadas manualmente por Integrative Genomics Viewer (Robinson et al., 2011). Debido al gran número de DNMs identificados, para la validación de las variantes por secuenciación Sanger, se seleccionaron al azar 51 variantes de novo de un solo nucleótido. La secuenciación Sanger se realizó con un analizador genético ABI PRISM 3130xl. Todos los DNM filtrados y revisados manualmente identificados por VarScan (N = 95) y por VarSeqTM (N = 84) fueron anotados utilizando ANNOVAR (Butkiewicz y Bush, 2016; Wang et al., 2010). Para el análisis de las interacciones entre proteínas, se utilizó el software STRING (Szklarczyk et al., 2017). Como en el caso del mapeo del exoma, las anotaciones se realizaron utilizando el genoma humano de referencia hg19.
La probabilidad de que una posición de llamada fuera un DNM en el trío se calculó independientemente para cada trío. Como se describe en una referencia anterior (Besenbacher et al, 2015), la tasa de novo por posición por generación (PPPG) se calculó de la siguiente manera:
donde f es el número de tríos y N es el número de sitios llamables, que potencialmente pueden ser identificados como sitios de novo para cada trío por separado, independientemente de la profundidad de secuenciación. Este número varía dependiendo del trío. ni es el número de DNMs identificados para el trío i. La probabilidad Pji (de novos ingle nucleotide) para el llamado sitio de nucleotide único j y la familia i para ser mutado se calculó como sigue:
La probabilidad Pji (de novo indel)para que el sitio llamado indel j y la familia i sean mutados se calculó como:
donde C, M y F significan descendiente, madre y padre, respectivamente, y Hetero, HomR y HomA denotan heterocigoto, homocigoto para la referencia y homocigoto para el alelo alternativo, respectivamente. La probabilidad Pij (de novo) se calculó con respecto a la cobertura de secuenciación. Los intervalos de confianza para las estimaciones de tasas se calcularon como para las proporciones binomiales. Para la estimación de la tasa de DNM y para otros cálculos, se utilizó el paquete R (versión 3.4.3) (R Core Team, 2013).
Con el fin de probar la hipótesis de que las variaciones en la tasa de DNM a través de diferentes regiones del genoma podrían ser explicadas por las características intrínsecas de la propia región genómica y la edad de los padres, se realizó un análisis de regresión lineal, para lo cual se llevó a cabo la anotación «secundaria» de cada DNM utilizando datos de los proyectos ENCODE (ENCODE Project Consortium, 2012) y LITGEN (LITGEN, 2011). En primer lugar, de acuerdo con un estudio anterior (Besenbacher et al., 2015), para recopilar registros relativos al paisaje genómico de los DNM identificados, se eligieron líneas celulares linfoblastoides (LCL y GM12878) (ENCODE Project Consortium, 2012). Se recogieron datos de:
(1) índices de expresión (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) en diferentes tejidos. Según la expresión de las regiones con DNMs se dividieron en posiciones con expresión específica y no específica;
(2) mediciones de los sitios de hipersensibilidad a la DNasa1 (DHS). El estado del DHS se asignó a 0 si estaba fuera del pico del DHS y a 1 si estaba dentro;
(3) mediciones del contexto de las islas CpG. Si el DNM estaba dentro de las islas CpG se asignó un estado de posición 1; si estaba fuera – 0;
(4) tres marcas de histonas (H3K27ac, H3K4me1, y H3K4me3) del proyecto ENCODE. Si el DNM estaba en la posición marcada con la histona se asignó con 1 y si no – 0;
(5) Los valores de conservación GERPP++ se recogieron utilizando la herramienta de anotación ANNOVAR. Según los valores de conservación las posiciones con DNMs se asignaron en posiciones conservadoras (puntuación GERP++ >12) y no conservadoras (puntuación GERP++ <12) (Davydov et al., 2010; ENCODE Project Consortium, 2012). A partir de los registros del cuestionario del proyecto LITGEN, se recogieron datos sobre la edad de los padres. Tras la recopilación de los parámetros para cada trío, se calculó el número de posiciones con cada parámetro. A continuación, se realizó un análisis de correlación seguido de un modelo de regresión lineal de la tasa de DNM y los parámetros.
Resultados
Tras el análisis de DNM, se identificó un número excepcionalmente alto de DNM para dos tríos (nº 4 y 21): 113 y 123 (mediante VarScan y VarSeqTM, respectivamente) y 16 (VarScan). Estos resultados nos llevaron a probar la paternidad biológica, que fue rechazada para el trío no. 4 y confirmada para el trío núm. 21. Así pues, los datos del trío n.º 4 se excluyeron del estudio. 4 fueron excluidos del estudio. En el conjunto final de 48 tríos, se identificaron 95 DNM en 34 tríos con el software VarScan y 84 DNM en 31 tríos con el software VarSeqTM (Figura 1). No se detectaron DNMs en 18 y 15 tríos por VarScan y VarSeqTM, respectivamente. De todos los DNMs identificados por ambos programas de software, sólo el 5,37% de los DNMs coincidieron (tres DNMs en los genes MEIS2, PGK1 y MT1B). Cada persona tenía 1,9 (software VarScan) y 1,7 (VarSeqTM) DNMs de media.
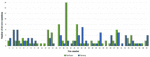
FIGURA 1. Comparación de las variantes de nucleótido único de novo identificadas por el software VarScan (azul) y VarSeqTM (verde).
El análisis de 95 DNMs que fueron identificados por el software VarScan mostró que 20 DNMs eran exónicos, incluyendo dos DNMs de ganancia de parada, siete DNMs sinónimos y 11 DNMs no sinónimos. Ochenta nuevas mutaciones identificadas por VarSeqTM eran exónicas, incluyendo 1 DNM de ganancia de parada y 78 DNM no sinónimas (Figura 2). La mayoría de los DNM identificados por VarScan se encontraban en los cromosomas 1, 2, 4 y 5, mientras que VarSeqTM identificó los DNM predominantemente en los cromosomas 2, 6, 7 y 11. El número de DNM identificados no se correlacionó con la densidad de genes en los cromosomas (R = 0,09, valor p = 0,65 para VarScan y R = 6,73, valor p = 0,51 para VarSeqTM) ni con el tamaño del cromosoma (Figura 3). Según ambos programas informáticos, los ratios de transiciones y transversiones fueron muy similares: 1,44 y 1,47, respectivamente (Figura 4). Sin embargo, se identificaron diferencias en las estructuras de las transiciones. En concreto, entre los DNM identificados por VarScan, había más cambios G/T y A/C, mientras que entre los DNM identificados por VarSeqTM, había más cambios A/T y G/C.
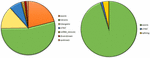
FIGURA 2. Composición de las mutaciones de novo (DNM) generadas por VarScan (a la izquierda) y por VarSeqTM (a la derecha).
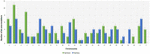
FIGURA 3. Distribución del número de variantes de novo por cromosoma según los datos generados por VarScan y VarSeqTM. Las barras verdes representan los DNMs identificados por el software VarScan, azul – por VarSeqTM. Las barras de error representan el error estándar de la media de los DNM para cada cromosoma.
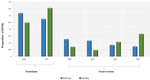
FIGURA 4. Los eventos moleculares que subyacen a las transiciones ocurren con mayor frecuencia que los que conducen a las transversiones, lo que resulta en una tasa ∼1,5 veces mayor de transiciones sobre transversiones en todo el exoma. Eventos de transición y transversión identificados por el software VarScan (verde) y VarSeqTM (azul). Las barras de error representan el error estándar de la media de DNMs.
Las tasas calculadas de mutaciones de novo de un solo nucleótido fueron de 2,4 × 10-8 PPPG (intervalo de confianza del 95% : 1,96 × 10-8-2,99 × 10-8) según VarSeqTM y de 274 × 10-8 por nucleótido por generación (IC del 95%: 2,24 × 10-8-3,35 × 10-8) según VarScan.
El algoritmo VarScan identificó tres indels de novo en tres tríos en los cromosomas 6 y 11. La tasa calculada de indels de novo en el genoma fue de 1,77 × 10-8 (IC del 95%: 6,03 × 10-9-5,2 × 10-8) PPPG. En particular, todos los indels de novo eran «reversibles», es decir, los padres tenían nuevas variantes en el genoma, y sus hijos tenían variantes de novo basadas en el genoma de referencia con el valor medio de 37,5 de profundidad de secuenciación y 50 de calidad del genotipo, respectivamente. Sin embargo, estos tres DNM no fueron seleccionados para la validación por el método de secuenciación de Sanger, por lo que sigue existiendo una probabilidad de sobreestimación de los indels de novo. Los indels de novo fueron C/T y A/G en el contexto de nucleótidos individuales.
El modelado de regresión lineal reveló que los sitios de hipersensibilidad a la DNAse 1, el contexto de las islas CpG, los valores de conservación GERPP++ y los niveles de expresión explicaron ∼68-93% de las tasas de DNM (Tabla 1). Ni los marcadores epigenéticos ni la edad paterna se correlacionaron significativamente con la tasa de DNM. Los modelos se establecieron a partir de los datos obtenidos de VarScan únicamente porque no había correlación entre los datos de VarSeqTM y las características intrínsecas de la propia región genómica.

TABLA 1. La regresión lineal de los sitios de hipersensibilidad de la ADNasaI, el contexto de las islas CpG, los valores de conservación de GERPP++ y el efecto del nivel de expresión sobre la tasa de DNMs.
Predicción funcional de las DNMs
Para evaluar qué mutaciones missense eran deletéreas y alteraban la función de la proteína afectada por tipo, se analizaron las puntuaciones categóricas predichas para el daño inducido por las DNMs. Se consideraron los siguientes 10 valores: polifenética HDIV y HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, codificación Fathmm-MKL y GERP++. Basándose en las puntuaciones de las predicciones, se seleccionaron cuatro DNMs identificados por VarScan que tenían seis o más predicciones dañinas o probablemente dañinas. Estos DNMs con ganancia de parada estaban en los genes MEIS2 y ULK4, mientras que los DNMs no sinónimos estaban en los genes MT1B y PGK1. Las proteínas codificadas por estos genes son importantes para el crecimiento neuronal, la endocitosis y la protección contra los efectos negativos de los metales pesados. Estas proteínas participan en la liberación del inhibidor de los vasos sanguíneos del tumor, la angiostatina, y en varias vías de señalización. No hubo conexiones entre las proteínas codificadas por estos genes (Figura 5).
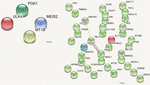
FIGURA 5. Interacciones proteína-proteína (Szklarczyk et al., 2017) en los genes que albergan DNMs. Los DNMs identificados por VarScan en genes que codifican proteínas están a la izquierda, los DNMs identificados por VarSeqTM – a la derecha. Las líneas de color indican una conexión entre proteínas.
Las mutaciones de novo identificadas por VarSeqTM se analizaron con más detalle si se predecía que eran perjudiciales o probablemente perjudiciales por al menos la mitad de las herramientas de predicción. Hubo 35 mutaciones puntuales (ver ??) en genes que codifican proteínas importantes para la remodelación de la cromatina, la regulación del citoesqueleto, el crecimiento y la viabilidad de las células, las vías de señalización citoplasmática y el inicio de las respuestas neuronales que desencadenan la percepción del olor.
Entre las proteínas codificadas por los genes afectados por los DNMs, sólo CLPTM1, ZNF547 y DMXL1 estaban conectados de alguna manera (Figura 5).
Discusión
En este estudio, realizamos un análisis exhaustivo de la distribución de los DNMs a través de diferentes regiones del exoma en la población lituana. En total, se detectaron 95 DNMs en 34 tríos y 84 DNMs en 31 tríos utilizando la tecnología de secuenciación SOLiD 5500 mediante los algoritmos VarScan y VarSeqTM, respectivamente. En primer lugar, nos gustaría señalar que elegimos VarScan para la llamada de DNMs porque de acuerdo con (Warden et al., 2014) este algoritmo resulta una lista de variantes, con alta concordancia (>97%) a las variantes de alta calidad llamadas por el GATK UnifiedGenotyper y HaplotypeCaller. Se eligió el software VarSeqTM porque es una herramienta ampliamente utilizada para el análisis de variantes tanto en investigaciones como en análisis clínicos. A pesar de que ambos algoritmos están diseñados para buscar DNMs en el exoma de la descendencia que no estaban presentes en ninguno de los padres, la concordancia entre los dos programas de software para el análisis de DNMs fue sólo del 5,37%. El algoritmo VarScan tuvo una mayor sensibilidad (5,42%) para la detección de DNM antes de la filtración que el algoritmo VarSeqTM (1,77%), por lo que sospechamos que ninguna herramienta tuvo éxito en la llamada de mutaciones debido a la alta sensibilidad que siempre estuvo acompañada de una baja especificidad. Por lo tanto, sugerimos que podría lograrse una mejora considerable de los resultados combinando la salida de diferentes herramientas (Sandmann et al., 2017).
Basado en los datos generados, la tasa estimada de DNM de un solo nucleótido fue de entre 2,4 × 10-8 y 2,74 × 10-8 y la de indels de novo fue de 1,77 × 10-8 PPPG, dependiendo del algoritmo utilizado. Nuestra tasa de DNM calculada fue mayor que la reportada en estudios anteriores (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015), en los que varió entre 1,2 × 10-8 y 1,5 × 10-8 PPPG. La mayor tasa de DNM en nuestro estudio era razonable porque nuestro estudio se basaba en datos de exomas. Además, los exomas presentan tasas de mutación significativamente más altas (en un 30%) que los genomas completos porque la composición de pares de bases del genoma completo es diferente a la de los exomas. En particular, los exomas tienen un contenido medio de GC de aproximadamente el 50%, mientras que el del genoma completo es de aproximadamente el 40% (Neale et al., 2012). Los CpGs metilados representan secuencias altamente mutables en los humanos debido a la desaminación espontánea de las bases de citosina (Neale et al., 2012). Según los estudios de genómica comparativa, se cree que las mayores tasas de mutación en las regiones ricas en CpGs evolucionaron alrededor de la época de la radiación de los mamíferos (Francioli et al., 2015). Durante la divergencia de las especies, las regiones exónicas ricas en CpG sufrieron mayores tasas de mutación en comparación con las del ADN no codificante y se convirtieron en regiones no codificantes. Por lo tanto, entonces el efecto del contenido de CpG disminuye con el tiempo, la tasa media de mutación disminuye hasta que alcanza el nivel presente en el ADN no codificante circundante (Subramanian y Kumar, 2003). Sin embargo, mientras que las secuencias en las regiones del genoma que evolucionan de forma neutra han tenido tiempo suficiente para equilibrarse con respecto a los contextos dinucleotídicos, la selección purificadora ha mantenido los CpGs hipermutables en las regiones funcionales (Subramanian y Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Por lo tanto, debido a que encontramos una tasa de DNM más alta que la reportada por otros estudios, especulamos que podría deberse, al menos parcialmente, al contexto de la secuencia local y/o a una posible presión de selección natural en el exoma. En consecuencia, se aplicó un modelo de regresión lineal, y encontramos que la hipersensibilidad a la DNAse 1, el contexto de las islas CpG, los valores de conservación GERPP++ y el nivel de expresión explicaban ∼68-93% de la tasa de DNM. Estos resultados indicaron que los DNM en el exoma se formaron independientemente de la conservación de las secuencias de ADN. Sin embargo, la tasa de DNM fue mayor en los genes cuyos productos eran inespecíficos y en las regiones de tipo promotor transcripcionalmente activas.
En contraste con los resultados de otros estudios (Wong et al., 2016; Sandmann et al., 2017), encontramos que la edad paterna no se correlacionó con la tasa de DNM. Estos resultados podrían explicarse por el hecho de que el conjunto de datos consistía en tríos con edades paternas similares y que solo se analizó una pequeña porción (∼1,5%) de todo el genoma. Sobre la base de estos parámetros, cada persona tenía sólo 1,9 (VarScan) o 1,7 (VarSeqTM) DNMs en promedio en comparación con 40-82 en todo el genoma (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al, 2016), mientras que el número de indels de novo en la secuencia codificante fue similar al identificado en (Front Line Genomics, 2017).
Los resultados de nuestro extenso análisis funcional de las anotaciones revelaron que de todos los DNMs identificados, 4 (VarScan) y 35 (VarSeqTM) variantes eran probablemente DNMs patógenos. La diferencia en el número de DNMs patógenos puede explicarse por el hecho de que, dependiendo del algoritmo utilizado para la identificación de DNMs, la proporción de DNMs en las secuencias codificantes difería significativamente. Por ejemplo, el 21,05% de los DNM identificados por el software VarScan eran exónicos, mientras que el 95,24% de los identificados por el software VarSeqTM eran exónicos. Estos DNM patógenos se encontraban en los genes que codifican proteínas esenciales para el modelado de la cromatina, la regulación del citoesqueleto, la modulación del crecimiento y la vitalidad celular, la función de las vías de señalización citoplasmática y el inicio de la respuesta neuronal. A pesar de que estos DNM se consideran patógenos, todos los individuos que participaron en la encuesta se identificaron como genéticamente «sanos». Por lo tanto, este resultado indicaba que, a pesar de la putativa patogenicidad de las DNM, los genomas en los que se localizaban las DNM obviamente toleraban dichos cambios, por lo que las manifestaciones de la enfermedad no solían ser pronunciadas. Según Szamecz et al. (2014), cuanto más a menudo ocurren los DNM en posiciones genéticas conservadas, más fuertes son los efectos de la selección natural sobre los cambios genéticos a través de mecanismos compensatorios de protección del genoma. Los efectos nocivos de las variantes pueden mitigarse de cuatro maneras. Algunos genes pueden tolerar las variantes truncadas de las proteínas porque sus efectos funcionales están enmascarados por la expresión incompleta, las variantes compensatorias o la escasa importancia funcional del truncamiento (Bartha et al., 2015). Por el contrario, los cambios génicos asociados a las DNM no sinónimas se compensan mediante el mecanismo de acumulación de mutaciones útiles en todo el genoma (Szamecz et al., 2014). Esto sugiere que en estos casos, las mutaciones patógenas no son lo suficientemente deletéreas como para reducir la aptitud media y, por lo tanto, persisten durante más tiempo en muchas generaciones siendo moldeadas por la selección natural.
En resumen, nuestro análisis de la distribución de DNMs y de su contexto genético y epigenético proporcionó conocimientos sobre la variación genética del genoma lituano. En base a estos hallazgos, estudios adicionales en grupos de pacientes con enfermedades genéticas pueden facilitar nuestra capacidad para distinguir ciertos DNMs patógenos de los DNMs de fondo tolerados y para identificar DNMs causales fiables. Sin embargo, la principal limitación de este estudio radica en que no hemos examinado la variación en las regiones genéticas no codificantes y reguladoras. Esta información podría contribuir a la elucidación de los posibles mecanismos de formación de los DNM que aún no están suficientemente claros.
Códigos de acceso
Los datos de las secuencias se han depositado en el Archivo Europeo de Nucleótidos (ENA), bajo el acceso PRJEB25864 (ERP107829).
Declaración ética
Este estudio se llevó a cabo de acuerdo con las recomendaciones de permiso, Comité Regional de Ética de Vilnius para la Investigación Biomédica. El protocolo fue aprobado por el Comité Regional de Ética de Vilnius para la Investigación Biomédica. Todos los sujetos dieron su consentimiento informado por escrito de acuerdo con la Declaración de Helsinki.
Contribuciones de los autores
LP realizó el análisis de los datos y preparó el manuscrito. AJ calculó la tasa de mutaciones de novo. La secuenciación de los exomas de los tríos fue realizada por LA e IK. VK fue el investigador principal.
Financiación
Este estudio fue apoyado por el Fondo Social Europeo bajo la medida de Subvención Global. Proyecto LITGEN no. VP1-3.1-ŠMM-07-K-01-013.
Declaración de conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un potencial conflicto de intereses.
Material complementario
El material complementario de este artículo puede encontrarse en línea en: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., y Hoischen, A. (2016). Nuevos conocimientos sobre la generación y el papel de las mutaciones de novo en la salud y la enfermedad. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). Las características de las variantes de truncamiento de proteínas heterocigotas en el genoma humano. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). Anotación funcional in silico de la variación genómica. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Los orígenes, patrones e implicaciones de la mutación espontánea humana. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identificación de una alta fracción del genoma humano que está bajo restricción selectiva usando GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Una enciclopedia integrada de elementos de ADN en el genoma humano. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., y Keightley, P. D. (2007). La distribución de los efectos de fitness de las nuevas mutaciones. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Revista Front Line Genomics número 14 – ASHG. Londres: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Efectos genéticos en la expresión génica a través de los tejidos humanos. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: descubrimiento de mutaciones somáticas y alteraciones del número de copias en el cáncer mediante secuenciación del exoma. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Tasa de mutaciones de novo y la importancia de la edad del padre para el riesgo de enfermedad. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Diferencias en la tasa de recombinación a pequeña escala entre sexos, poblaciones e individuos. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ‘t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). La secuenciación del transcriptoma y del genoma descubre la variación funcional en los seres humanos. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Disponible en: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patrones y tasas de mutaciones exónicas de novo en los trastornos del espectro autista. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., y Waxman, D. (2018). Qué es la adaptación y cómo debe medirse? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). Un lenguaje y entorno para la computación estadística. Viena: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Visor de genómica integrativa. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluación de herramientas de llamada de variantes para datos de secuenciación de próxima generación no coincidentes. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., and Sunyaev, S. (2008). Hypermutable non-synonymous sites are under stronger negative selection. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Las sustituciones neutrales se producen a un ritmo más rápido en los exones que en el ADN no codificante en los genomas de los primates. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). El paisaje genómico de la evolución compensatoria Be. El paisaje genómico de la evolución compensatoria. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). La base de datos STRING en 2017: redes de asociación proteína-proteína de calidad controlada, hechas ampliamente accesibles. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: anotación funcional de variantes genéticas a partir de datos de secuenciación de próxima generación. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Comparación detallada de dos paquetes populares de llamadas de variantes para estudios de exomas y exones dirigidos. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Nuevas observaciones sobre el efecto de la edad materna en las mutaciones de novo en la línea germinal. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar
.