Alexei Drummond, Andrew Rambaut, Remco Bouckaert y Walter Xie
1 Introducción
Este tutorial introduce el software BEAST para el análisis evolutivo bayesiano a través de un sencillo tutorial. El tutorial involucra la co-estimación de una filogenia de genes y los tiempos de divergencia asociados en presencia de información de calibración de la evidencia fósil.
Necesitará el siguiente software a su disposición:
- BEAST – este paquete contiene el programa BEAST, BEAUti, TreeAnnotator y otros programas de utilidad. Este tutorial está escrito para BEAST v2.2.x, que tiene soporte para múltiples particiones. Está disponible para su descarga en http://www.beast2.org/.
- Tracer – este programa se utiliza para explorar la salida de BEAST (y otros programas Bayesianos MCMC). Resume gráfica y cuantitativamente las distribuciones de los parámetros continuos y proporciona información de diagnóstico. En el momento de escribir este artículo, la versión actual es la v1.6. Está disponible para su descarga en
http://tree.bio.ed.ac.uk/software/. - FigTree – es una aplicación para visualizar e imprimir filogenias moleculares, en particular las obtenidas mediante BEAST. En el momento de escribir este artículo, la versión actual es la v1.4.2. Está disponible para su descarga en http://tree.bio.ed.ac.uk/software/.
Este tutorial le guiará a través del análisis de un alineamiento de secuencias muestreadas de doce especies de primates (ver Figura 1). El objetivo es estimar la filogenia, la tasa de evolución en cada linaje y las edades de las divergenciasancestrales no calibradas.
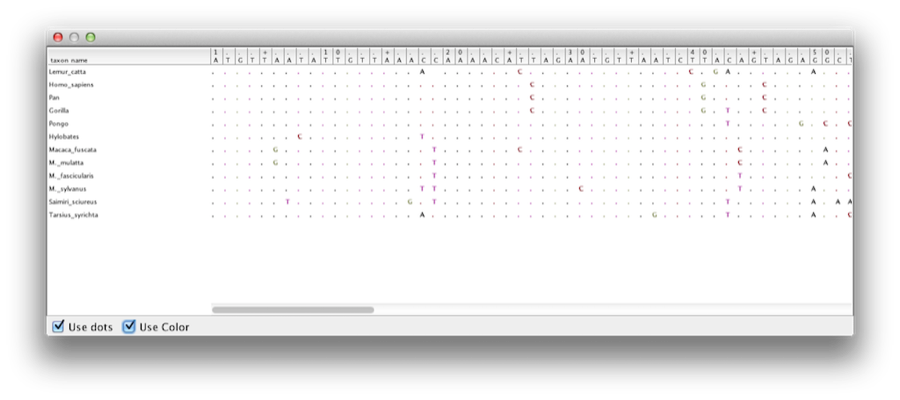
Figura 1: Parte del alineamiento para primates.
El primer paso será convertir un archivo NEXUS con un bloque DATA o CHARACTERS en un archivo de entrada BEAST XML. Esto se hace utilizando el programa BEAUti (que significa Bayesian Evolutionary Analysis Utility). Se trata de un programa fácil de usar para establecer el modelo evolutivo y las opciones para el análisis MCMC. El segundo paso consiste en ejecutar BEAST utilizando el archivo de entrada generado por BEAUTi, que contiene los datos, el modelo y la configuración del análisis. El último paso es explorar la salida de BEAST para diagnosticar problemas y resumir los resultados.
2 BEAUti
El programa BEAUti es un programa fácil de usar para establecer los parámetros del modelo para BEAST. Ejecute BEAUti haciendo doble clic en su icono. Una vez ejecutado, BEAUti tendrá un aspecto similar independientemente del sistema informático en el que se ejecute. Para este tutorial, se utiliza la versión de Mac OS X en las figuras, pero las versiones de Linux y Windows tendrán el mismo diseño y funcionalidad.
2.1 Carga del archivo NEXUS
Para cargar una alineación en formato NEXUS, simplemente seleccione la opción Importar Alineación… del menú Archivo, o arrastre el archivo al centro del panel de Particiones.
El archivo de ejemplo llamado primate-mtDNA.nex está disponible en el directorio examples/nexus/ para Mac y Linux y examples/nexus/ para Windows dentro del directorio donde se instaló BEAST.Este archivo contiene un alineamiento de secuencias de 12 especies de primates.
Se abrirá una ventana Add Partition (Figura 2) si el paquete relacionado está instalado. Si está utilizando BEAST 2 «puro», puede pasar al siguiente apartado. De lo contrario, seleccione Añadir Alineación y haga clic en Aceptar para continuar.
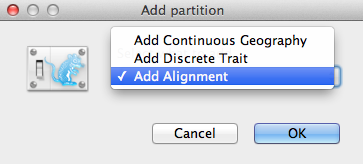
Figura 2: Ventana de Añadir Partición (Sólo aparece si los paquetes relacionados están instalados).
Si hay alguna superposición de codificación en las particiones, la ventana de mensaje de advertencia (Figura 3) aparecerá. Lea y haga clic en OK para continuar.
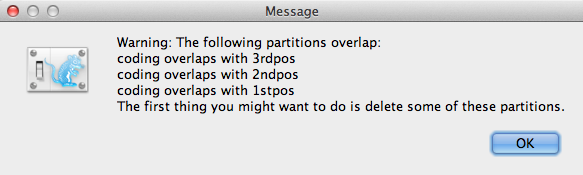
Figura 3: Ventana de mensaje de advertencia (Sólo aparece si hay solapamientos de codificación en las particiones).
Una vez cargado, se muestran cinco particiones de caracteres en el panel principal (Figura 4). La alineación se divide en una parte codificante de la proteína y una parte no codificante,y la parte codificante se divide en las posiciones de codones 1, 2 y 3. Debe eliminar la partición «codificante» antes de continuar con el siguiente paso, ya que se refiere a los mismos nucleótidos que las particiones «1stpos», «2ndpos» y «3rdpos». Para eliminar la partición ‘coding’ seleccione la fila y haga clic en el botón ‘-‘ en la parte inferior de la tabla. Puede ver la alineación haciendo doble clic en la partición.
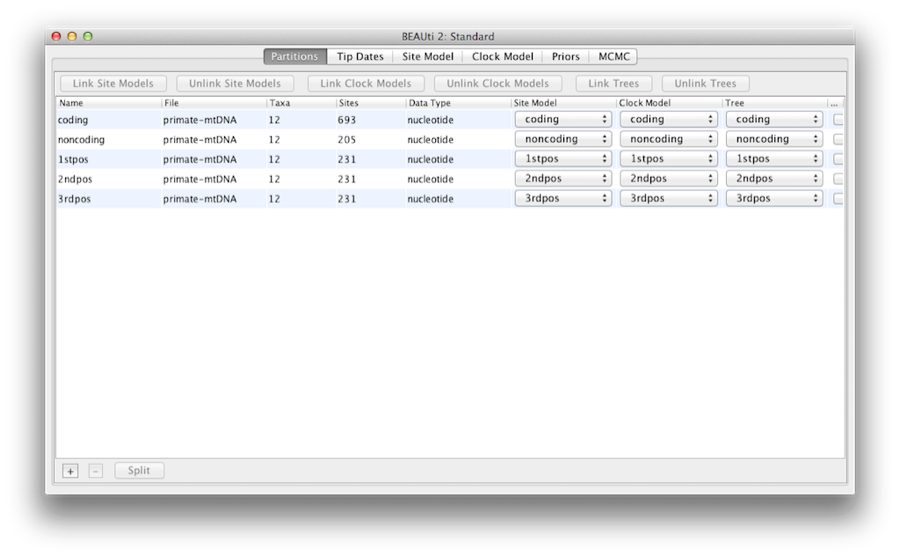
Figura 4: Una captura de pantalla de la pestaña de datos en BEAUti. Esta y todas las siguientes capturas de pantalla fueron tomadas en un ordenador Apple con Mac OS X y tendrán un aspecto ligeramente diferente en otros sistemas operativos.
Enlazar/desenlazar modelos de partición
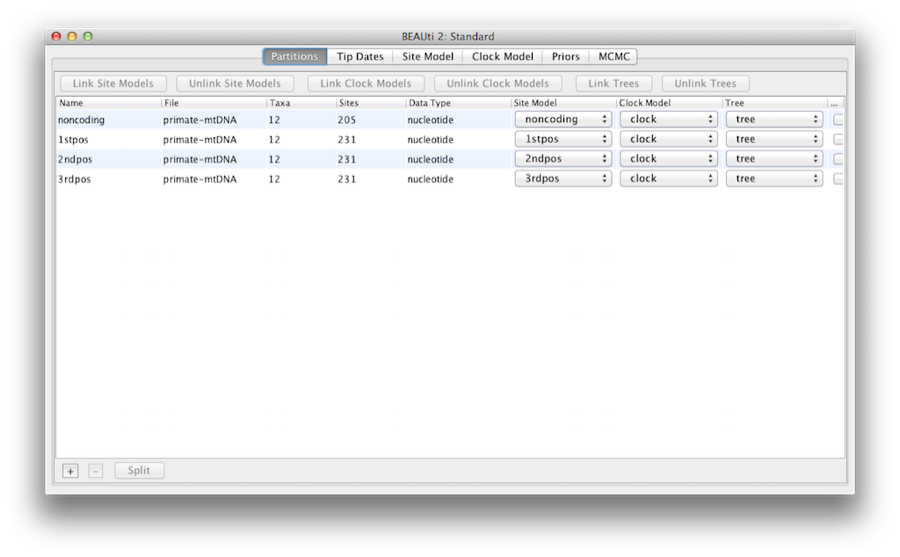
Figura 5: Una captura de pantalla de la pestaña Particiones en BEAUti después de enlazar y renombrar el modelo de reloj y el árbol.
Dado que las secuencias están enlazadas (es decir, todas provienen del genoma mitocondrial que no se cree que sufra recombinación en aves y mamíferos) comparten la misma ascendencia, por lo que las particiones deberían compartir el mismo árbol temporal en el modelo. En aras de la simplicidad, también supondremos que las particiones comparten la misma tasa evolutiva para cada rama y, por tanto, el mismo «modelo de reloj».
Entonces, en este punto necesitaremos vincular el modelo de reloj y el árbol. En el panel de Particiones, seleccione las cuatro particiones de la tabla (o ninguna, por defecto todas las particiones están afectadas) y haga clic en el botón Enlazar Árboles y luego en el botón Enlazar Modelos de Reloj (ver Figura 5). A continuación, haga clic en el primer menú desplegable de la columna Modelo de reloj y cambie el nombre del modelo de reloj compartido a ‘reloj’. Del mismo modo, cambie el nombre del árbol compartido a «árbol». Esto hará que las siguientes opciones y los archivos de registro generados sean más fáciles de leer.
2.2 Configuración del modelo de sustitución
El siguiente paso es configurar el modelo de sustitución. A continuación, seleccione la pestaña Site Models en la parte superior de la ventana principal (nos saltamos la pestaña Tip Dates ya que todos los taxones proceden de muestras contemporáneas). Esto revelará los ajustes del modelo evolutivo para BEAST. Las opciones disponibles dependen de si los datos son nucleótidos o aminoácidos, datos binarios o datos generales. Los ajustes que aparecerán después de cargar el alineamiento de nucleótidos de primates serán los valores por defecto para los datos de nucleótidos, por lo que tenemos que hacer algunos cambios.
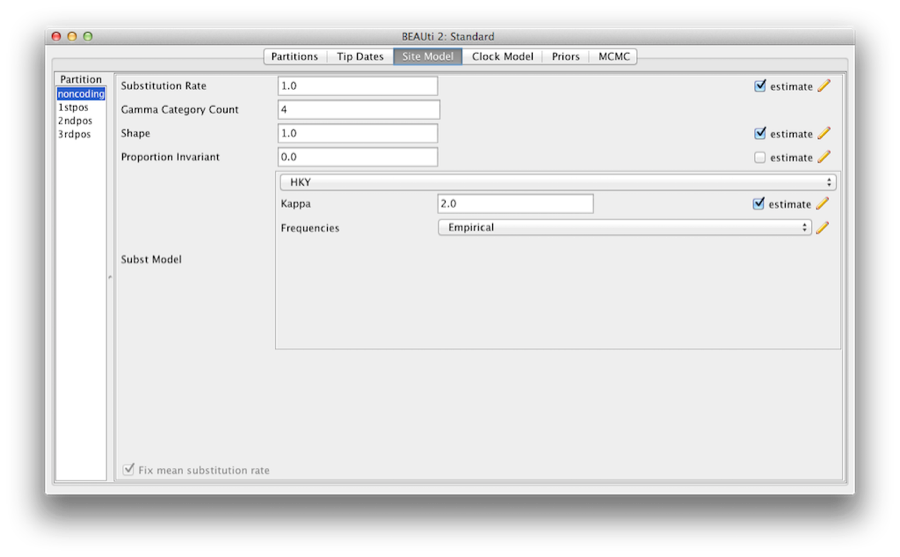
Figura 6: Una captura de pantalla de la pestaña de modelo de sitio en BEAUti.
La mayoría de los modelos deben ser familiares para usted. En primer lugar, establezca el recuento de la categoría gamma a 4 y, a continuación, marque la casilla «estimación» para el parámetro Shape. Esto permitirá modelar la variación de la tasa entre los sitios de cada partición. Tenga en cuenta que de 4 a 6 categorías funcionan suficientemente bien para la mayoría de los conjuntos de datos, mientras que tener más categorías lleva más tiempo de cálculo para poco beneficio añadido. Dejamos la entrada Proportion Invariant en cero.
A continuación, seleccione HKY en el menú desplegable Subst Model. Idealmente, debería seleccionarse un modelo de sustitución que se ajustara mejor a los datos para cada partición, pero aquí, en aras de la simplicidad, utilizamos HKY para todas las particiones. Además, seleccione Empírico en el menú desplegable Frecuencias. Esto fijará las frecuencias a las proporciones observadas en los datos (para cada partición individualmente, una vez que desligamos los modelos de sitio). Este enfoque significa que podemos obtener un buen ajuste a los datos sin estimar explícitamente estos parámetros. Lo hacemos aquí simplemente para hacer los archivos de registro un poco más cortos y más legibles en partes posteriores del ejercicio.
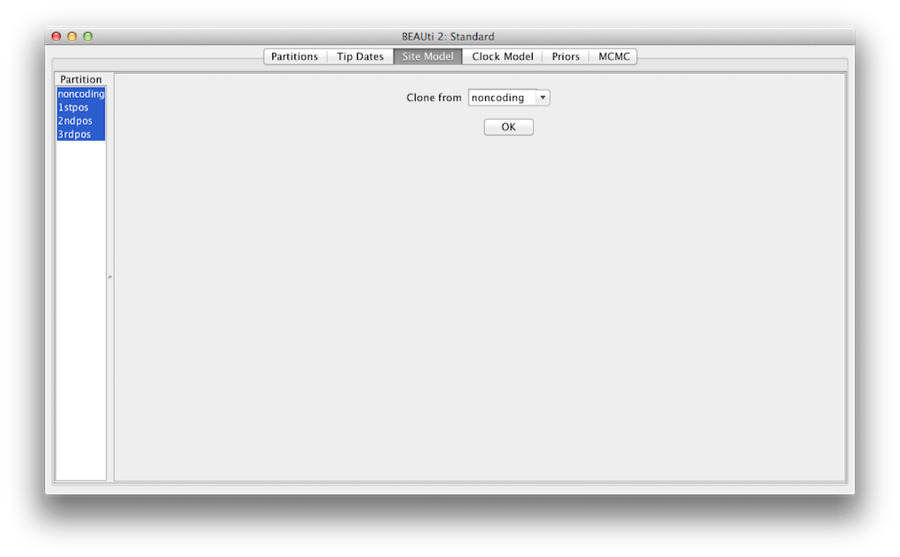
Figura 7: configuración de clones de un modelo de sitio a otros.
Por último, marque la casilla «estimar» para el parámetro Tasa de sustitución y seleccione la casilla Fijar tasa de mutación media. Esto permitirá que las particiones individuales tengan sus tasas relativas estimadas para desvincular los modelos de sitio (Figura 6).
Por último, mantenga la tecla ‘shift’ para seleccionar todos los modelos de sitio en el lado izquierdo, y haga clic en OK para clonar el ajuste de no codificación en 1stpos, 2ndpos y 3rdpos (Figura 7). Revise cada modelo de sitio, como puede ver, sus configuraciones son las mismas ahora.
2.3 Configuración del modelo de reloj
El siguiente paso es seleccionar la pestaña Clock Models en la parte superior de la ventana principal. Aquí es donde seleccionamos el modelo de reloj molecular. Para este ejercicio vamos a dejar la selección en el valor por defecto de un reloj molecular estricto, porque estos datos son muy parecidos a un reloj, y no necesita la variación de la tasa entre las ramas para ser incluido en el modelo.
Para comprobar la similitud con el reloj, puede (i) ejecutar el análisis con un modelo de reloj relajado y comprobar cuánta variación entre las tasas está implícita en los datos (véase el coeficiente de variación para más información), o (ii) realizar una comparación de modelos entre un reloj estricto y uno relajado utilizando el muestreo de rutas, o (iii) utilizar un modelo de reloj local aleatorio que considere explícitamente si cada rama del árbol necesita su propia tasa de ramificación.
2.4 Priores
La pestaña Priores permite especificar los priores para cada parámetro del modelo. Las selecciones del modelo realizadas en las pestañas del modelo del sitio y del modelo del reloj, resultan en la inclusión de varios parámetros en el modelo, y estos se muestran en la pestaña de priores (ver Figura 8).
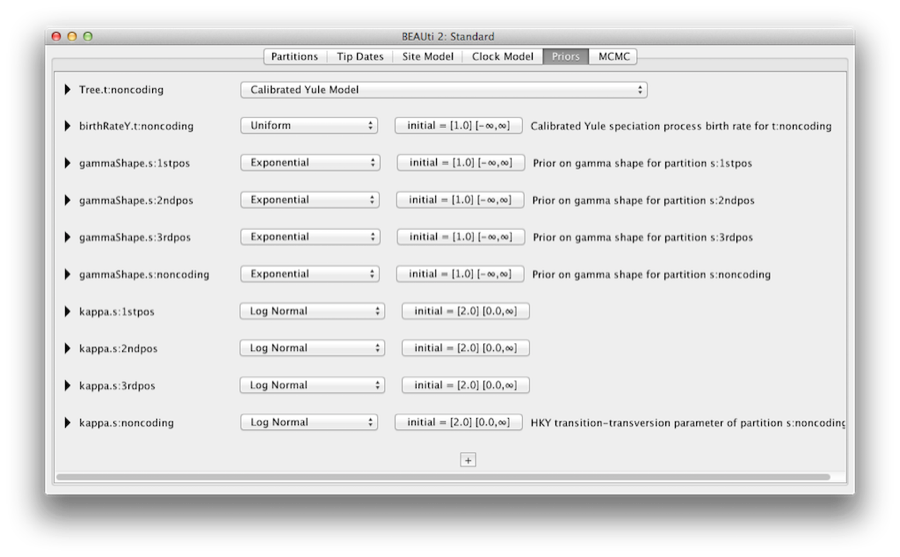
Figura 8: Una captura de pantalla de la pestaña de Priores en BEAUti.
Aquí también especificamos que deseamos utilizar el modelo calibrado de Yule como la prioridad del árbol. El modelo de Yule es un modelo simple de especificación que generalmente es más apropiado cuando se consideran secuencias de diferentes especies. Seleccione el modelo de Yule calibrado en el menú desplegable Tree prior.
2.4.1 Definición del nodo de calibración
Ahora tenemos que especificar una distribución a priori en el nodo calibrado, basada en nuestros conocimientos fósiles. Esto se conoce como calibrar nuestro árbol. Para definir una prioridad adicional, pulse el pequeño botón + debajo de la lista de prioridades. Si no está visible en su vista, desplácese por el panel hasta la parte inferior para encontrar el botón +. Verá un diálogo que le permite definir un subconjunto de taxones en el árbol filogenético. Una vez que haya creado un conjunto de taxones, podrá añadir la información de calibración de su ancestro común más reciente (MRCA) más adelante.
Nombre el conjunto de taxones rellenando la entrada de la etiqueta del conjunto de taxones. Llámelo human-chimp, ya que contendrá los taxones de Homo sapiens y Pan. En la lista de abajo verá los taxones disponibles. Seleccione cada uno de los dos taxones a su vez y pulse el botón de flecha > >. (Como se trata de un nodo calibrado que se utilizará junto con el prior de Yule calibrado, debe aplicarse la monofilia, así que seleccione la casilla marcada como Monofilético. Esto restringirá la topología del árbol para que la agrupación humano-chimpancé se mantenga monofilética durante el curso del análisis MCMC.
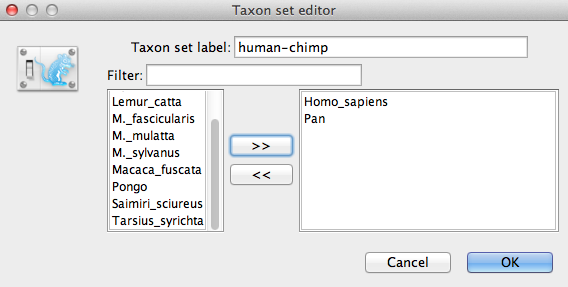
Figura 9: Editor del conjunto de taxones en BEAUti.
Para codificar la información de calibración necesitamos especificar una distribución para el MRCA del chimpancé-humano.Seleccione la distribución Log-normal en el menú desplegable a la derecha del recién añadido chimpancé-humano.prior. Haga clic en el triángulo negro y aparecerá un gráfico de la función de densidad de probabilidad, junto con los parámetros de la distribución logarítmica normal. Vamos a establecer M=1,78 y S=0,085, lo que especificará una distribución centrada en unos 6 millones de años con una desviación estándar de unos 0,5 millones de años. Esto dará un rango central de probabilidad del 95% que cubre de 5 a 7 Mya. Esto corresponde aproximadamente a la estimación de consenso actual de la fecha del ancestro común más reciente de los humanos y los chimpancés (Figura 10).
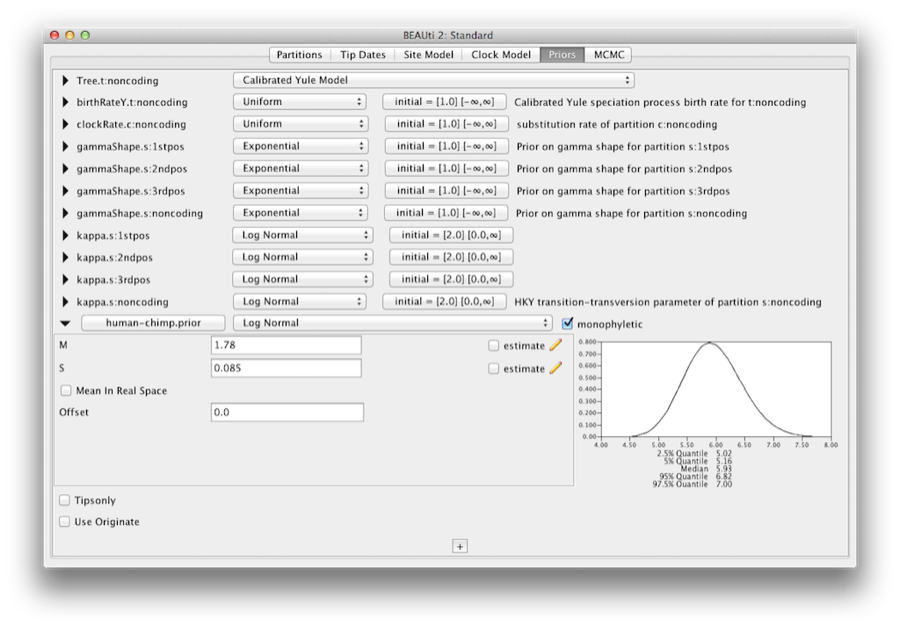
Figura 10: Captura de pantalla de las opciones de calibración a priori en el panel Priors de BEAUti.
Debemos convencernos de que las priorizaciones mostradas en el panel de priorizaciones reflejan realmente la información previa que tenemos sobre los parámetros del modelo. Por último, también especificaremos algunas priores difusas «no informativas» pero adecuadas sobre la tasa global del reloj molecular (clockRate) y la tasa de especiación (birthRateY) del árbol de Yule a priori. Para cada una de ellas, seleccione Gamma en el menú desplegable y, con el botón de la flecha, amplíe la vista para ver los parámetros de la prioridad Gamma. Tanto para la tasa de reloj como para la tasa de nacimiento de Yule, establezca el parámetro Alfa (forma) en 0,001 y el parámetro Beta (escala) en 1000 (Figura 11).
Por defecto, cada uno de los parámetros de la forma gamma tiene una distribución priordistributiva exponencial con una media de 1. Esto implica (véase la Figura 3.7) que esperamos alguna variación. Por defecto, los parámetros kappa para el modelo HKY tienen una distribución a priori lognormal(1,1.25), que coincide ampliamente con la evidencia empírica sobre el rango de valores realistas para los sesgos de transición/transversión. Estas prioridades por defecto se mantienen ya que son adecuadas para este análisis en particular.
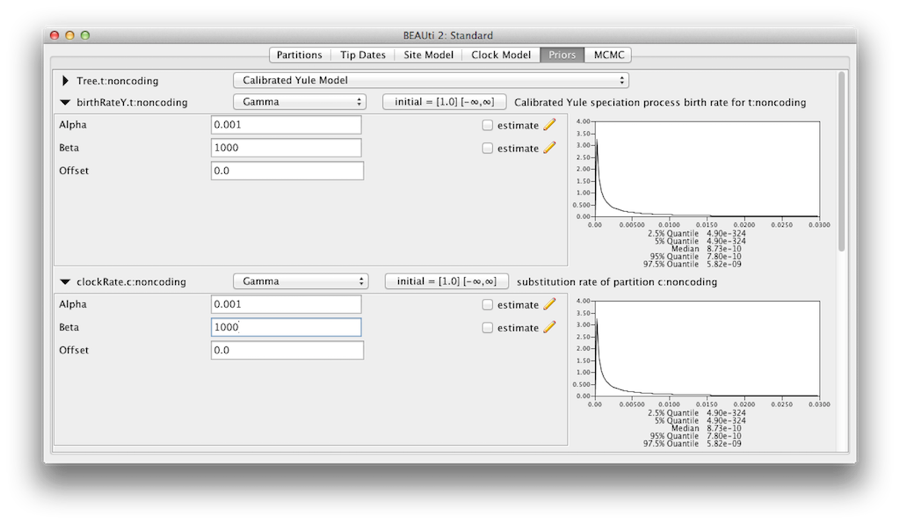
Figura 11: Prioridad Gamma.
2.5 Configuración de las opciones MCMC
La siguiente pestaña, MCMC, proporciona más ajustes generales para controlar la duración de la ejecución MCMC y los nombres de los archivos.
En primer lugar tenemos la longitud de la cadena. Este es el número de pasos que el MCMC hará en la cadena antes de terminar. La longitud de la cadena depende del tamaño del conjunto de datos, la complejidad del modelo y la calidad de la respuesta requerida. El valor por defecto de 10.000.000 es totalmente arbitrario y debería ajustarse según el tamaño de su conjunto de datos. Para este conjunto de datos vamos a establecer la longitud de la cadena a 6.000.000 ya que esto se ejecutará razonablemente rápido en la mayoría de los ordenadores modernos (unos pocos minutos).
El campo Almacenar cada determina la frecuencia con la que el estado se almacena en el archivo. Almacenar el estado periódicamente es útil para situaciones en las que el entorno informático no es muy fiable y una ejecución de BEAST puede ser interrumpida. Tener una copia almacenada del estado reciente permite reanudar la cadena en lugar de reiniciar desde el principio, por lo que no es necesario volver a pasar por el burn-in.El campo Pre Burnin especifica el número de muestras que no se registran al principio del análisis. Dejamos los campos Store Every y Pre Burnin con sus valores por defecto. Debajo de ellos se encuentran los detalles de los archivos de registro. Cada uno de ellos puede ampliarse haciendo clic en el triángulo negro.
Las siguientes opciones especifican la frecuencia con la que los valores de los parámetros de la cadena de Markov deben mostrarse en la pantalla y registrarse en el archivo de registro.La salida de la pantalla es simplemente para monitorear el progreso de los programas, por lo que puede establecerse en cualquier valor (aunque si se establece demasiado pequeño, la cantidad de información que se muestra en la pantalla realmente ralentizará el programa). Para el archivo de registro, el valor debe establecerse en relación con la longitud total de la cadena. Si el muestreo es demasiado frecuente, los archivos serán muy grandes y la precisión del análisis no se verá afectada. Si el muestreo es demasiado infrecuente, el archivo de registro no registrará suficiente información sobre las distribuciones de los parámetros. Es probable que desee almacenar no más de 10.000 muestras, por lo que esto debe ser no menos de la longitud de la cadena / 10.000.
Para este ejercicio vamos a establecer el registro de rastreo y la frecuencia de registro de árbol a 1.000 y el registro de pantalla a 10.000.También especifique Primates.log como el nombre del archivo de registro de rastreo y Primates.trees como el nombre del archivo de registro de árbol.Asegúrese de que el nombre del archivo del registro de la pantalla se deja vacío, o el registro de la pantalla no se escribirá en la pantalla.
- Si está utilizando el sistema operativo Windows, entonces le sugerimos que añada el sufijo .txt a ambos (así, Primates.log.txt y Primates.trees.txt) para que Windows los reconozca como archivos de texto.
2.6 Generación del archivo XML de BEAST
Ahora estamos listos para crear el archivo XML de BEAST. Para ello, seleccione la opción Guardar del menú Archivo. Marque los valores predeterminados y guarde el archivo con un nombre apropiado (normalmente el nombre del archivo termina con .xml, por ejemplo, Primates.xml).
3 Ejecutar BEAST
Figura 12: Una captura de pantalla de BEAST.
Ahora ejecute BEAST y cuando le pida un archivo de entrada, proporcione su archivo XML recién creado como entrada. BEAST se ejecutará hasta que termine de mostrar la información en la pantalla. Los archivos de resultados reales se guardan en el disco en la misma ubicación que el archivo de entrada. La salida a la pantalla tendrá un aspecto similar al siguiente:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Nótese que hay alguna información útil al principio sobre las alineaciones y qué probabilidades de árbol se utilizan. Además, todas las citas relevantes para el análisis se mencionan al principio de la ejecución, lo que puede copiarse fácilmente en los manuscritos que informan sobre el análisis. A continuación, se informa de la cadena, lo que proporciona información en tiempo real sobre el progreso de la cadena.
Al final, se imprime un análisis de operadores, que enumera todos los operadores utilizados en el análisis junto con la frecuencia con la que el operador fue probado, aceptado y rechazado (ver columnas #total, #aceptar y #rechazar respectivamente). La tasa de aceptación es la proporción de veces que se acepta un operador cuando se selecciona para hacer una propuesta. En general, una tasa de aceptación elevada, por ejemplo superior a 0,5, indica que las propuestas son conservadoras y no exploran el espacio de parámetros de forma eficaz. Por otro lado, una tasa de aceptación baja indica que las propuestas son demasiado agresivas y casi siempre dan lugar a un estado que se rechaza debido a su bajo nivel posterior. Una tasa de aceptación de 0,234 es el objetivo (basado en pruebas muy limitadas proporcionadas por ) para muchos (pero no todos) los operadores implementados en BEAST.
Algunos operadores tienen un parámetro de ajuste, por ejemplo el factor de escala del parámetro ascale. Si la tasa de aceptación final no está cerca del objetivo, BEAST sugerirá un nuevo valor para el parámetro de ajuste, que se imprime en el análisis del operador. En este caso, todas las tasas de aceptación son buenas para los operadores que tienen parámetros de ajuste. Los operadores sin parámetros de sintonía incluyen los operadores wideexchange y Wilson-Balding para este análisis. Ambos operadores intentan cambiar la topología del árbol con pasos grandes, pero como los datos apoyan una topología única de forma abrumadora, estas propuestas radicales son casi siempre rechazadas.
4 Análisis de los resultados
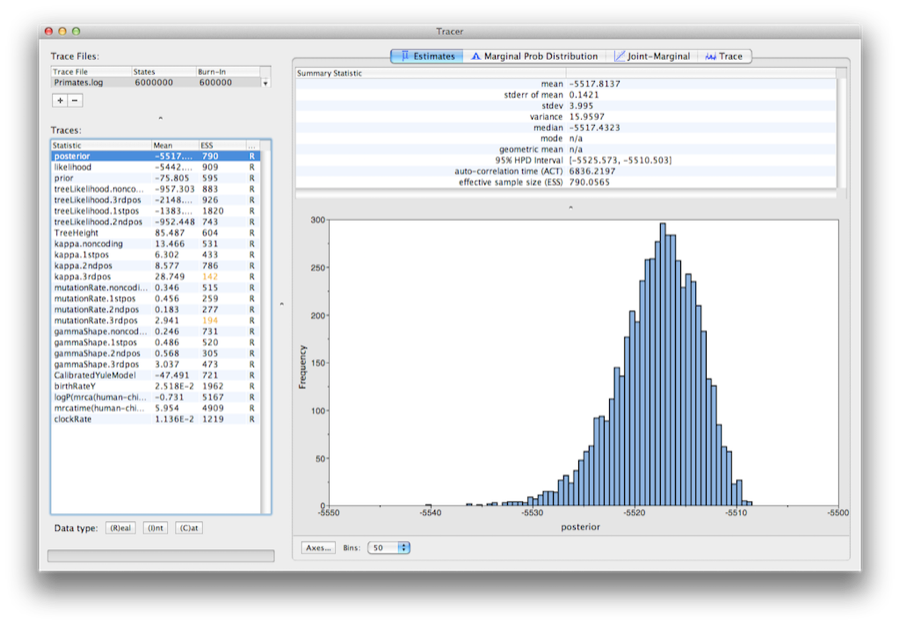
Figura 13: Captura de pantalla de Tracer v1.6.
Ejecute el programa llamado Tracer para analizar la salida de BEAST. Cuando se abra la ventana principal, elija Import Trace File… en el menú File y seleccione el archivo que BEAST ha creado llamado Primates.log (Figura 13).
Recuerde que MCMC es un algoritmo estocástico por lo que los números reales no serán exactamente los mismos que los representados en la figura.
En la parte izquierda hay una lista de las diferentes cantidades que BEAST ha registrado en el archivo. Hay trazas para el posterior (esto es el logaritmo natural del producto de la probabilidad del árbol y la densidad previa), y los parámetros continuos. Al seleccionar una traza a la izquierda, aparecen los análisis de esta traza a la derecha, dependiendo de la pestaña seleccionada. Cuando se abre por primera vez, se selecciona la traza ‘posterior’ y se muestran varios estadísticos de esta traza en la pestaña Estimaciones.En la parte superior derecha de la ventana hay una tabla de estadísticos calculados para la traza seleccionada.
Seleccione el parámetro clockRate en la lista de la izquierda para ver la tasa media de evolución (promediada en todo el árbol y en todos los sitios). Tracer trazará un histograma (marginal posterior) para la estadística seleccionada y también le dará estadísticas resumidas como la media y la mediana. El 95% HPD significa intervalo de mayor densidad posterior y representa el intervalo más compacto del parámetro seleccionado que contiene el 95% de la probabilidad posterior. Se puede considerar como un análogo bayesiano de un intervalo de confianza. El parámetro TreeHeight da la distribución posterior marginal de la edad de la raíz de todo el árbol.
Seleccione el parámetro TreeHeight y luego haga Ctrl-clic en mrcatime(human-chimp) (Comando-clic en Mac OS X). Esto mostrará una visualización de la edad de la raíz y la calibración MRCA que especificamos anteriormente en BEAUti. Puede comprobar que la divergencia que utilizamos para calibrar el árbol(mrcatime(human-chimp)) tiene una distribución posterior que coincide con la distribución a priori que especificamos (Figura 14).
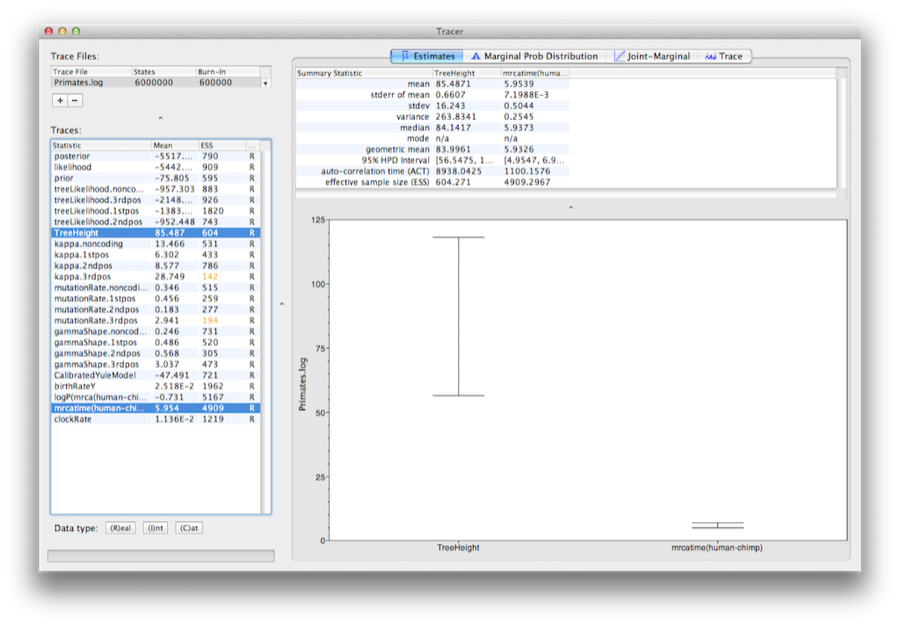
Figura 14: Captura de pantalla de los intervalos HPD al 95% de la altura de la raíz y el MRCA especificado por el usuario (human-chimp) en Tracer.
5 Estimaciones posteriores marginales
Para mostrar las tasas relativas de las cuatro particiones, seleccione el parámetro mutationRate para cada una de las cuatro particiones y seleccione la pestaña de densidad marginal en Tracer.La figura 15 muestra las densidades marginales para las tasas de sustitución relativas. El gráfico muestra que las posiciones del codón 1 y 2 tienen tasas sustancialmente diferentes (0,456 frente a 0,183) y ambas son mucho más lentas que la posición del codón 3, con una tasa relativa de 2,941. La partición no codificante tiene una tasa intermedia entre las posiciones de codones 1 y 2 (0,346). En conjunto, este resultado sugiere una fuerte selección purificadora tanto en las regiones codificadoras como en las no codificadoras del alineamiento.
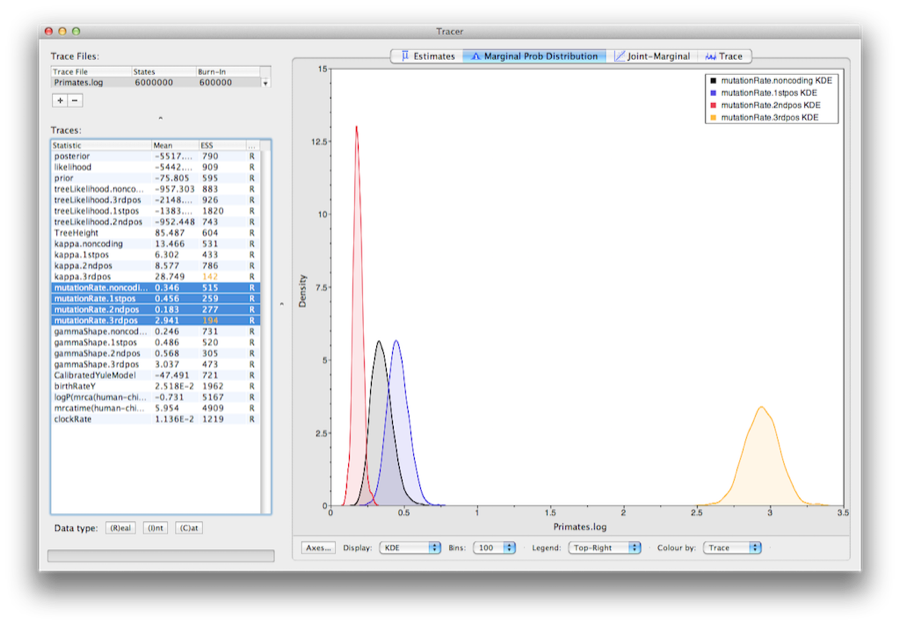
Figura 15: Captura de pantalla de las densidades marginales posteriores de las tasas de sustitución relativas de las cuatro particiones (en relación con la tasa media ponderada por sitio).
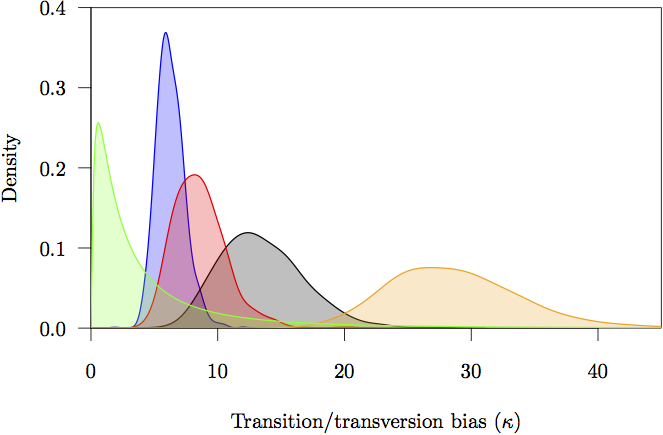
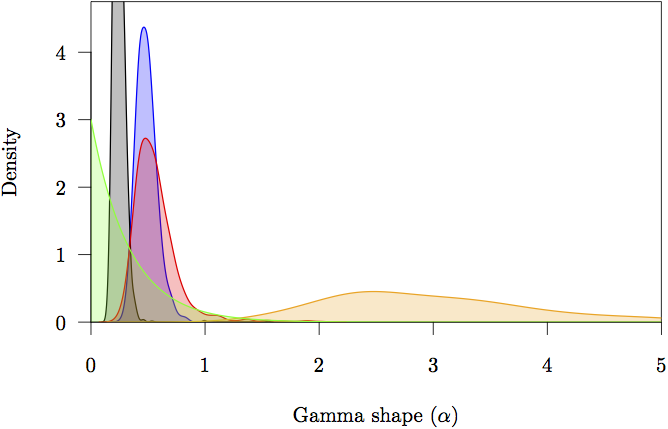
Figura 16: Las densidades marginales a priori y a posteriori de los parámetros de forma (α). La prioridad está en gris. También se muestra la densidad posterior estimada para cada partición: posiciones de codones no codificantes (naranja) y primera (rojo), segunda (verde) y tercera (azul).
Figura 17: Las densidades marginales a priori y a posteriori para los parámetros de sesgo de transición/transversión (κ). La prioridad está en gris. También se muestra la estimación de la densidad posterior para cada partición: posiciones de codones no codificantes (naranja) y primera (rojo), segunda (verde) y tercera (azul).
Preguntas
¿Cuál es la tasa estimada de evolución molecular para este árbol de genes (incluya el intervalo HPD del 95%)?
¿Qué fuentes de error incluye esta estimación?
¿Cuál es la edad de la raíz del árbol (dé la media y el intervalo HPD del 95%)?
6 Obtención de una estimación del árbol filogenético
BEAST también produce una muestra posterior de árboles temporales filogenéticos junto con su muestra de estimaciones de parámetros. Estos deben ser resumidos utilizando el programa TreeAnnotator. Éste tomará el conjunto de árboles y encontrará el mejor soportado. A continuación, anotará este árbol resumen representativo con las edades medias de todos los nodos y los correspondientes rangos de HPD del 95%. También calculará la probabilidad de clado posterior para cada nodo. Ejecute el programa TreeAnnotator y configúrelo como se muestra en la Figura 18.
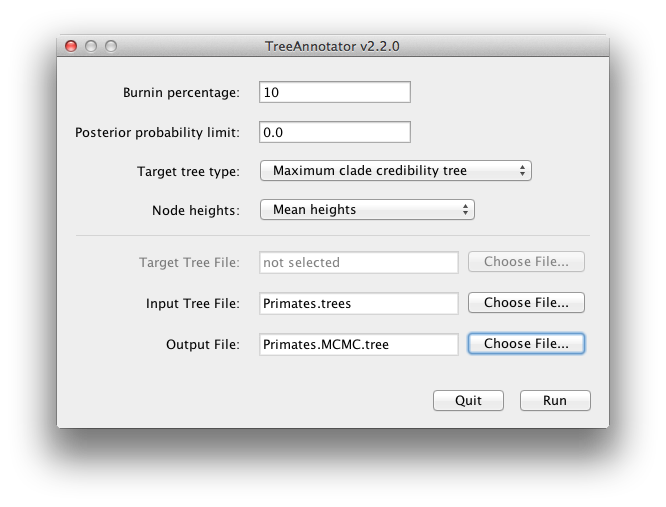
Figura 18: Captura de pantalla de TreeAnnotator.
El burnin es el número de árboles a eliminar desde el inicio de la muestra. A diferencia de Tracer, que especifica el número de pasos como burnin, en TreeAnnotator es necesario especificar el número real de árboles. Para esta ejecución, se ha especificado una longitud de cadena de 6.000.000 de pasos muestreando cada 1.000 pasos. Por lo tanto, el archivo de árboles contendrá 6.000 árboles y, por lo tanto, deberá especificar un 10% de burnin en el campo de texto superior.
La opción Límite de probabilidad posterior especifica un límite tal que si un nodo se encuentra con menos de esta frecuencia en la muestra de árboles (es decir, tiene una probabilidad posterior inferior a este límite), no se anotará. El valor predeterminado de 0,5 significa que sólo se anotarán los nodos que se encuentren en la mayoría de los árboles. Establezca este valor en cero para anotar todos los nodos.
El tipo de árbol objetivo especifica la topología del árbol que se anotará. Puede elegir un árbol específico de un archivo o pedir a TreeAnnotator que encuentre un árbol en su muestra.La opción por defecto, Maximum clade credibility tree, encuentra el árbol con el mayor producto de la probabilidad posterior de todos sus nodos.
Para las alturas de los nodos, el valor por defecto es Common Ancestor Heights, que calcula la altura de un nodo como la media del tiempo MRCA de todos los pares de nodos en el clado. En el caso de árboles con una gran incertidumbre en la topología y, por tanto, con muchos clados con bajo soporte, algunos otros métodos pueden dar lugar a árboles con longitudes de rama negativas. En este análisis, el soporte de todos los clados en el árbol resumen es muy alto, por lo que esto no es un problema en este caso.Elija las alturas medias para las alturas de los nodos. Esto establece las alturas (edades) de cada nodo en el árbol a la altura media a través de la muestra completa de árboles para ese clado.
Para el archivo de entrada, seleccione el archivo de árboles que BEAST creó y seleccione un archivo para la salida (aquí lo llamamos Primates.MCC.tree). Ahora pulse Ejecutar y espere a que el programa termine.
7 Visualización de la estimación del árbol
Por último, podemos visualizar el árbol en otro programa llamado FigTree. Ejecute este programa y abra el archivo Primates.MCC.tree mediante el comando Abrir del menú Archivo. Ahora puede intentar seleccionar algunas de las opciones del panel de control de la izquierda. En primer lugar, gaste la opción Trees en el panel, y marque Order nodes y elija Ordering by decreasing. Pruebe a seleccionar Barras de nodos para obtener barras de error de la edad de los nodos. También active la opción Branch Labels y seleccione posterior para que se muestre la probabilidad posterior de cada nodo. Si utiliza un modelo de reloj no estricto, entonces en Apariencia también puede decirle a FigTree que coloree las ramas según la tasa.Debería terminar con algo similar a la Figura 19.
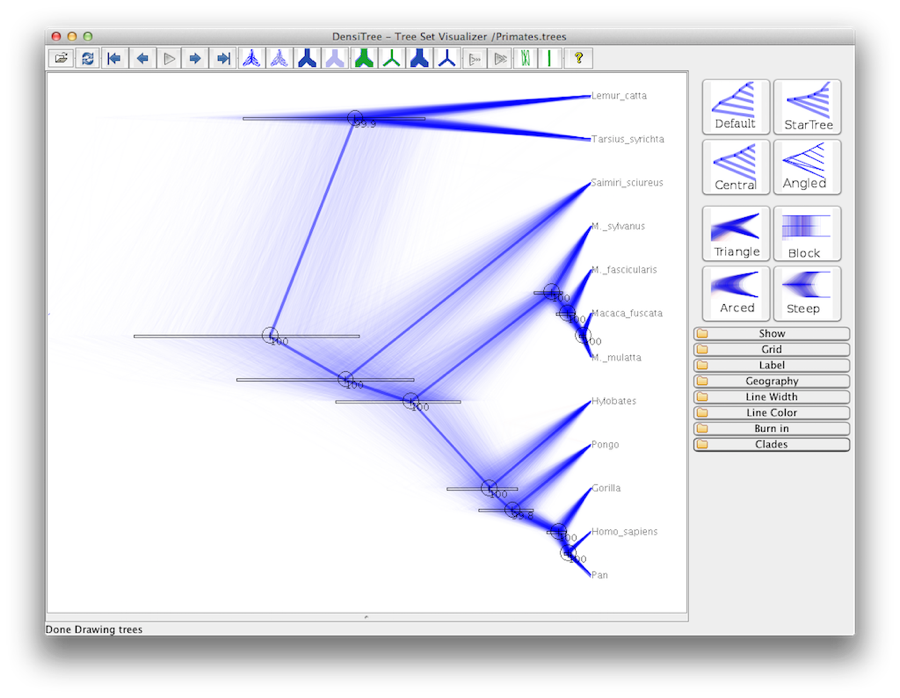
Figura 19: Una captura de pantalla de FigTree y DensiTree.
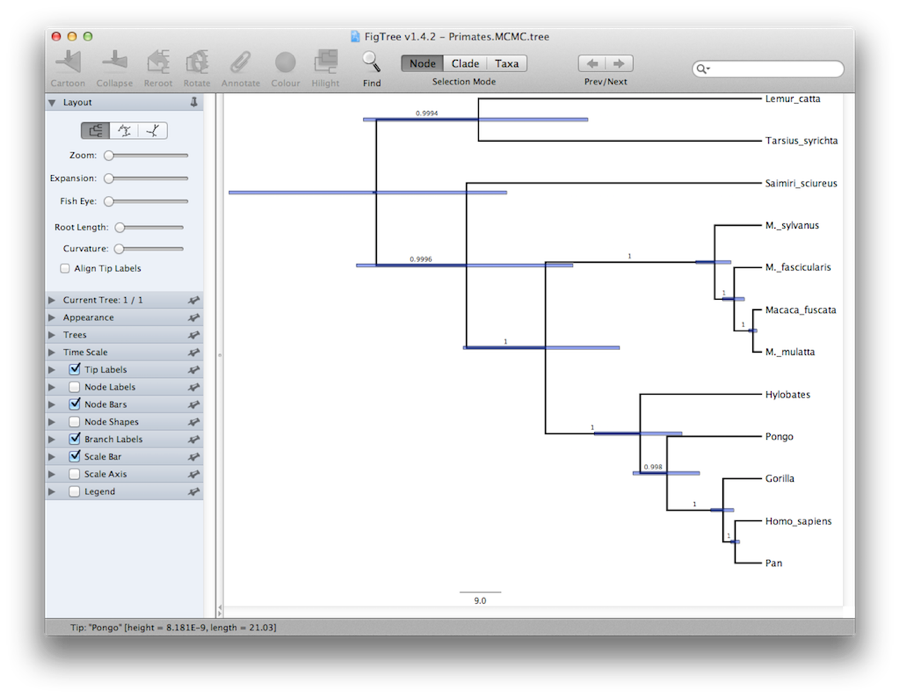
Una visión alternativa del árbol puede hacerse con DensiTree, que forma parte de Beast 2. La ventaja de DensiTree es que es capaz de visualizar tanto la incertidumbre en las alturas de los nodos como la incertidumbre en la topología.Para este conjunto de datos en particular, la topología dominante está presente en más del 99% de las muestras. Por lo tanto, concluimos que este análisis resulta en un consenso muy alto en la topología (Figura 19).
Preguntas
- ¿La tasa de evolución difiere sustancialmente entre los diferentes linajes del árbol?
- DensiTree tiene una barra de clados (Menú Ventana/Ver barra de herramientas de clados) para mostrar información sobre los clados.
¿Cuál es el soporte del clado?
- Puede navegar a través de las topologías en DensiTree utilizando el menú Examinar.La topología más popular tiene un soporte de más del 99%.
¿Cuál es el soporte de la segunda topología más popular?
- Bajo el menú de ayuda, DensiTree muestra alguna información.
¿Cuántas topologías hay en el conjunto de árboles?
8 Comparación de sus resultados con el prior
Es una buena idea volver a ejecutar el análisis mientras se muestrea desde el prior para asegurarse de que las interacciones entre los priores no están afectando a su información previa. La interacción entre prioritarios puede ser problemática, especialmente cuando se utilizan calibraciones, ya que significa poner múltiples prioritarios en el árbol.
Usando BEAUti, configure el mismo análisis pero en las opciones MCMC, seleccione la opción Sample from prior only. Esto le permitirá visualizar la distribución a priori completa en ausencia de sus datos de secuencia. Resuma los árboles de la distribución a priori completa y compare el resumen con el árbol de resumen posterior.
La estimación del tiempo de divergencia utilizando la «datación de nodos» del tipo descrito en este capítulo se ha aplicado para responder a una variedad de preguntas diferentes en ecología y evolución. Por ejemplo, la datación por nodos con fósiles se utilizó para determinar la diversidad de especies de las cícadas, analizar el ritmo de evolución de las plantas con flores e investigar los orígenes de las cianobacterias de los desiertos cálidos y fríos.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond y Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), no. 1, 114. A Gelman, G Roberts y W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled y Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little y S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian, y Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith y Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008),no. 5898, 86-89.
Este documento fue traducido de LATEX porHEVEA.