John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1Biomedisiinisten tieteiden tiedekunta, Kent State University, Ohio, USA
Biologisten tieteiden osasto, Kent State University, Ohio, USA
Abstract
Kasvava näyttö korostaa tyypin II diabeteksen (T2D) ja Alzheimerin taudin (AD) läheistä yhteyttä. On tärkeää, että näillä kahdella sairaudella on useita patologisia yhtäläisyyksiä, kuten amyloidin kertyminen, oksidatiivinen stressi, tulehdus ja solukuolema. Toistaiseksi Alzheimerin taudin ja T2D:n lääkehoitoja ei ole, ja näiden sairauksien uusien hoitomuotojen löytäminen ja kehittäminen on erittäin tärkeää. Useat ihmisillä ja jyrsijöillä tehdyt tutkimukset ovat osoittaneet, että aineenvaihduntahormonien lisääminen on erittäin arvokasta kognitiivisten toimintojen ja yleisen metabolisen terveyden parantamiseksi sekä T2D:ssä että Alzheimerin taudissa. Haimahormoni amyliini on noussut esiin ratkaisevana osatekijänä sekä T2D:n että Alzheimerin taudin etiologiassa, vaikka amyliinin tarkkaa roolia näissä sairauksissa ei vielä tunneta hyvin. Tässä tarkastelemme kriittisesti nykyistä kirjallisuutta, jossa hyödynnetään ihmisen amyliiniä tai sen synteettistä analogia, pramlintidiä, sekä amyliinireseptoriantagonisteja Alzheimerin taudin hoidossa.
Esittely
Alzheimerin tauti (AD) on etenevä, invalidisoiva hermoston rappeutumissairaus, jolle on ominaista amyloidi-beeta- (Aβ-) plakkien ja hyperfosforyloituneesta tau1:stä koostuvien neurofibrillaaristen säkkipilareiden (tangles) kertyminen. Näiden patologisten peptidien kertyminen aiheuttaa puutteita toimeenpanevissa toiminnoissa, kuten oppimisessa ja muistissa, mielialassa, vaikutuksissa jne., ja aiheuttaa huomattavaa taakkaa potilaalle ja hoitajille. Alzheimerin taudin esiintyvyys lisääntyy hälyttävää vauhtia Yhdysvalloissa: vuonna 2017 Alzheimerin tautia sairasti arviolta 5,5 miljoonaa amerikkalaista, ja määrän odotetaan kolminkertaistuvan vuoteen 2050 mennessä2. Lisäksi Alzheimerin tautia sairastavien potilaiden hoidon ja hoivan kustannukset ovat tällä hetkellä yli 200 miljardia dollaria vuodessa, ja niiden odotetaan vain kasvavan3. Vaikka Alzheimerin tauti on selvästi valtava ongelma Yhdysvalloissa ja sen ulkopuolella, hoitovaihtoehdot ovat edelleen hyvin rajalliset4. Monia lääkekokeita on tehty monilla erilaisilla kohdennetuilla lähestymistavoilla, mutta tällä hetkellä FDA:n hyväksymiä Alzheimerin taudin hoitoon tarkoitettuja lääkkeitä on vain kuusi, ja ne ovat vain oireenmukaisia hoitoja5, 6. Tähän mennessä suurin osa kehitetyistä farmakologisista aineista on kohdistunut nimenomaan Aβ- tai tau-patologiaan, mutta yksikään niistä ei ole onnistunut poistamaan tai estämään patologiaa4. Näin ollen on perustavanlaatuinen tarve kehittää käyttökelpoisia terapeuttisia ja ennaltaehkäiseviä hoitoja Alzheimerin tautiin.
Ikään liittyvä (sporadinen) Alzheimerin tauti on monimutkainen monitekijäinen sairaus, jolla on lukuisia geneettisiä ja ympäristöön liittyviä vaikutteita. Ympäristöllä ja elintavoilla on suuri merkitys sporadisen Alzheimerin taudin kehittymisessä; sellaiset tekijät kuin ruokavalio7-9, liikalihavuus8-10, metabolinen oireyhtymä7, tyypin II diabetes (T2D)9, 11 ja sydän- ja verisuonisairaudet12 ovat kaikki olleet osallisina Alzheimerin taudin synnyssä. On ratkaisevan tärkeää, että lihavuuden ja diabeteksen määrä kasvaa nopeasti samanaikaisesti Alzheimerin taudin kanssa12, 13. Vaikka lihavuuden ja Alzheimerin taudin välinen suhde on jokseenkin epäselvä, on näyttöä siitä, että keski-ikäisen lihavuudella on merkitystä Alzheimerin taudin kehittymisessä10. Vielä tärkeämpää on, että lihavuuteen liittyy yleisesti useita muita sairauksia, kuten sydän- ja verisuonitaudit, verenpainetauti, dyslipidemia, T2D, aivohalvaus jne.14. T2D:n esiintyvyys lisääntyy nopeasti, ja CDC:n arvion mukaan noin 30,3 miljoonalla ihmisellä (1 aikuinen 10:stä) Yhdysvalloissa on diabetes ja huikealla 84,1 miljoonalla (1 aikuinen 3:sta) on esidiabetes, ja suurin osa heistä ei ole tietoinen sairaudestaan. Lisäksi liikunnan laajamittaisen vähenemisen ja samanaikaisen ravinnonsaannin ja huonon ruokavalion lisääntymisen vuoksi liikalihavuuden, T2D:n, metabolisen oireyhtymän ja sydän- ja verisuonitautien määrän ehdotetaan vain kasvavan arviolta 600 miljoonaan T2D-tapaukseen maailmanlaajuisesti vuoteen 2035 mennessä15.
Todisteet, joiden mukaan aineenvaihdunnan toiminta ja sairaudet ovat osallisena kognitiivisen heikkenemisen ja ikääntymisen prosessissa, ovat huomattavat16, 17. Esimerkiksi noin 70 prosenttia T2D-diagnoosin saaneista henkilöistä raportoi kognitiivisesta heikentymisestä, ja huomattava osa T2D-potilaista sairastuu myöhemmin dementiaan16, 18-21. Vähintään viisi vuotta T2D:tä sairastaneilla henkilöillä on huomattavasti suurempi riski sairastua Alzheimerin tautiin kuin niillä, jotka ovat sairastaneet T2D:tä alle viisi vuotta17. Yhdessä nämä tiedot viittaavat siihen, että T2D:n yleistyminen väestössä saattaa osaltaan vaikuttaa Alzheimerin taudin yleistymiseen.
T2D:lle on aluksi ominaista korkea veren glukoosipitoisuus ja korkea insuliinipitoisuus, mikä johtaa hyperinsulinemiaan; on tärkeää, että amyliini, haiman β-saarekesolujen tuottama pieni aineenvaihduntahormoni, on yhdessä insuliinin kanssa, ja se erittyy yhdessä insuliinin kanssa, ja näin ollen se on T2D:ssä ylituotettu22. On tärkeää, että sekä T2D:ssä että Alzheimerin taudissa on useita patologisia piirteitä: 1) aivojen heikentynyt aineenvaihdunta ja metabolinen hormoniresistenssi 2) amyloidipatologia 3) oksidatiivinen stressi (OS) ja tulehdus. Krooninen, hyperinsulinemia ja hyperamylinemia johtavat useisiin fysiologisiin ongelmiin: krooninen hyperinsulinemia johtaa järjestelmän insuliiniresistenssiin22, heikentyneeseen insuliinin kulkeutumiseen veri-aivoesteen (BBB)23, 24 läpi ja siten heikentyneeseen insuliinin signalointiin aivoissa25. Insuliinisignaloinnin menetys aivoissa liittyy useisiin Alzheimerin tautiin liittyviin patologisiin piirteisiin, kuten lisääntyneeseen Aβ:n tuotantoon, tau-fosforylaatioon ja neuroinflammaatioon.
Lisäksi amyliinilla on samankaltaisia patologisia piirteitä kuin Aβ:llä suurina pitoisuuksina26 , ja se saattaa olla näiden kahden taudin yhteinen reitti. Amyliinifibrillejä on esimerkiksi löydetty 95 prosentilla T2D-potilaista27-29 haimasta, ja ne aiheuttavat useita fysiologisia häiriöitä, kuten poikkeavaa Ca2+ -tuloa, lisääntynyttä pro-inflammatoristen sytokiinien eritystä30,31 ja lopulta β-saarekesolujen menetystä32. Lisäksi amyliini läpäisee helposti BBB:n ja muodostaa aivoissa amyliinifibrillejä sekä Aβ:n kanssa sekoittuneita plakkeja, ja se saattaa olla vastuussa Alzheimerin taudin kaltaisesta patologiasta ja Aβ:n leviämisestä T2D:ssä33-35. Amyliinin tiedetään vaikuttavan pitkäaikaispotentiaatioon (LTP) hippokampuksessa, ja sillä voi olla luontainen vaikutus aivojen kognitiivisiin toimintoihin36-39. On kuitenkin edelleen epäselvää, onko amyliini myrkyllinen loukkaus näissä sairauksissa vai edistääkö sen toiminnallinen menetys aggregaation tai myöhäisvaiheen β-solukadon kautta T2D:ssä Alzheimerin taudin kehittymistä.
Amyliinin signaloinnin kahtiajako
Amyliinireseptorin (AMYR) ja amyliinisignaalin osallisuudesta T2D:n ja Alzheimerin taudin etenemisessä ja etiologiassa keskustellaan edelleen paljon. Tämän suhteen selvittämiseen tähtäävä tutkimuskokonaisuus laajenee nopeasti. Kaikki asiaankuuluvat tutkimukset ovat johdonmukaisesti osoittaneet, että amyliinisignaloinnin modulointi vaikuttaa AD:hen liittyvään patologiaan. Tämän suhteen luonnetta ei kuitenkaan ole vielä selvitetty konkreettisesti. Useat ryhmät ovat tuottaneet vakuuttavia tietoja, jotka viittaavat siihen, että amyliinisignaalien välityksellä voidaan ehkäistä Alzheimerin tautiin liittyvää patologiaa ja kognitiivisia puutteita sekä in vivo että in vitro40-44. Tärkeää on, että pramlintidi, joka on amyliinin rekombinantti, ei-aggregoituva muoto, jota käytetään yhdessä insuliinihoitojen kanssa diabeteksen hoidossa ja joka parantaa glykeemistä säätelyä, vähentää ruumiinpainoa ja vähentää seerumin OS-merkkiaineita45-47 , vaikuttaa lupaavalta myös AD:n hoitona. Tähän mennessä ei kuitenkaan ole tehty kliinisiä tutkimuksia, joissa amyliiniä tai pramlintiidiä olisi pyritty hyödyntämään terapeuttisena aineena dementian hoidossa. Jyrsijöillä tehdyistä tutkimuksista saadut selkeät todisteet viittaavat siihen, että krooninen hoito joko ihmisen amyliinilla tai pramlintidilla tuottaa vahvaa terapeuttista hyötyä Alzheimerin tautiin liittyvän patologian vähentämisessä; amyliini/pramlintidilisä vähentää liukoisen Aβ:n pitoisuuksia, plakkitaakkaa, tau-fosforylaatiota, neuroinflammaatiota ja OS:ää samalla kun se myös parantaa kognitiota40-42,44. Edellä mainitut tiedot viittaavat siihen, että synnynnäisen amyliinisignaalin menetys keskushermostossa aggregaation vuoksi aiheuttaa lisääntyneen riskin AD:n kehittymiselle, ja sitä käsitellään yksityiskohtaisemmin artikkelissa Grizzanti ym. 201848.
Tutkimukset osoittavat sitä vastoin myös, että ihmisen amyliinilla ja Aβ:llä on samanlaisia myrkyllisiä vaikutuksia ja että näitä myrkyllisiä vaikutuksia voidaan lievittää AMYR-antagonistilla36-39,49. Tiedot osoittavat esimerkiksi, että in vivo -hoito AMYR-antagonisteilla tuottaa hyvin samanlaisia fysiologisia hyötyjä kuin amyliini- tai pramlintidihoito. TgCRND8-AD-hiirten hoito AMYR-antagonistilla AC253 tai sen syklisellä vastineella cAC253 vähentää neuroinflammatiota, liukoisen Aβ:n pitoisuuksia ja plakkitaakkaa ja parantaa samalla kognitiota50. Vastaavasti in vitro/ex-vivo -tutkimukset osoittavat, että pieni annos ihmisen amyliiniä tai Aβ:tä aiheuttaa häiriöitä LTP:ssä ja että AC253 tai pramlintidi estävät nämä puutteet38,39. Suuremmat annokset ihmisen amyliiniä tai amyliini-oligomeerejä yhdistetään kontrolloimattomaan Ca2+-tulvaan, joka on vahvasti yhteydessä solukuolemaan26,32. Yhdessä nämä tiedot tukevat amyliini-oligomeerien toksista toimintaa ja siten AMYR-salpauksen mahdollista terapeuttista mekanismia. Toiset ovat sitä vastoin osoittaneet, että amyliinin suotuisat vaikutukset voidaan estää AC25341:llä. Näin ollen amyliinin hoidon tai eston terapeuttinen potentiaali on edelleen epäselvä ja korostaa amyloidien monimutkaista ja kaksijakoista luonnetta aivoissa ja periferiassa.
Piecing Together the Puzzle
Nykykirjallisuudessa on useita aukkoja, jotka on täytettävä, jotta amyliinitarinasta saataisiin kattavampi kuva: 1) synnynnäisen amyliinijärjestelmän luonne ja amyliinisignalointi aivoissa 2) Aβ:n ja pramlintidin signalointikyvyt kolmen tärkeimmän AMYR- ja sukulaisreseptorin kautta 3) terapeuttiset mekanismit, joilla amyliini/pramlintidin tai AMYR:n esto välittyy. Ensinnäkin mielenkiintoiset uudet tiedot osoittavat, että AMYR ei osallistu ainoastaan signalointiin vaan myös ligandin kuljetukseen BBB:n läpi. AMYR on heterodimeerinen reseptori, joka koostuu kalsitoniinireseptorista ja reseptoriaktiivisuutta muokkaavasta proteiinista (1-3)51 . Tätä varten kalsitoniinireseptorin (AMYR:n avainkomponentti) 50 %:n globaali tyrmäys vähensi merkittävästi aivoista löytyvän AC253:n määrää50 , mikä osoittaa, että BBB:ssä sijaitsevat AMYR:t osallistuvat näiden ligandien kuljettamiseen aivoihin ja että ne voivat myös osallistua amyliinin ja pramlintidin kuljettamiseen aivoihin tai aivoista ulos. Näiden BBB-kuljetusmekanismien olemassaolo viittaa siihen, että amyliinilla on todennäköisesti synnynnäinen fysiologinen tehtävä aivoissa, koska sen kulkeutumista aivoihin valvotaan tiukasti. On kuitenkin edelleen epäselvää, miten amyliinin signalointi tai sen puute johtaa Alzheimerin taudin patologisiin piirteisiin ja onko AMYR se väline, jonka kautta Aβ välittää toksisia vaikutuksiaan.
Aβ:n ja AMYR:n välisestä suhteesta on ristiriitaista näyttöä. Vaikka useat tutkimukset osoittavat selvästi, että ihmisen amyliinilla ja Aβ:llä on samanlaiset vaikutukset LTP:hen keskushermostossa ja että AMYR-estäjien käyttö parantaa näitä haitallisia vaikutuksia36-39, muut todisteet viittaavat siihen, että Aβ (1-42) ei kykene signaloimaan minkään AMYR:n kautta herättääkseen minkäänlaista cAMP-vastetta useilla eri pitoisuuksilla52. On mahdollista, että Aβ aktivoi erilaisia signalointikaskadeja vuorovaikutuksen kautta AMYR:n kanssa tai toimii yksinkertaisesti inerttinä kilpailevana inhibiittorina, mutta tätä ei ole vielä osoitettu.
Lisäksi erillinen tutkimus osoitti, että oligomeerinen amyliini välittää toksiset vaikutuksensa suoraan AMYR:n kautta ja epäsuorasti TRPV4:n, epäselektiivisen kationikanavan, kautta26. Ihmisen amyliinin pienet pitoisuudet aiheuttavat Ca2+-vasteen, joka välittyy sen natiivireseptorin kautta. Suuremmissa pitoisuuksissa ihmisen amyliini muodostaa kuitenkin oligomeerejä ja aktivoi poikkeavaa signalointia, joka johtaa TRVP4-kanavien aktivoitumiseen ja mahdollistaa kationien, erityisesti Ca2+:n, hallitsemattoman sisäänvirtauksen. AMYR:n ja TRPV4:n farmakologinen salpaaminen osoittaa, että molemmat reseptorit ovat välttämättömiä, jotta ihmisen oligomeerinen amyliini voi aiheuttaa myrkyllisiä Ca2+ -vaikutuksiaan26. Näin ollen on todennäköistä, että Aβ välittää toksiset vaikutuksensa AMYR:ään samalla tavalla, vaikka näitä tietoja ei vielä olekaan. Hallitsematon Ca2+ -tulva liittyy moniin patologisiin ilmiöihin, kuten hallitsemattomaan vesikkelien vapautumiseen, OS:n ja mitokondrioiden toimintahäiriöihin, apoptoosiin jne. Tätä varten on todennäköistä, että solujen toimintahäiriöt ja AD:n kaltaisen patologian kehittyminen, joka johtuu myrkyllisestä amyloidisignaalista, välittyy sekä AMYR:n että TRPV4:n kautta. Sellaisenaan on tarpeen erottaa ne signalointikaskadit, jotka moduloivat AMYR:n ja TRVP4:n välistä suhdetta. Lisäksi on perusteltua tehdä farmakologisia kokeita, joissa käytetään Aβ:tä ja pramlintidia laajalla annosvalikoimalla, jotta voidaan määrittää Aβ:n ja pramlintidin vaikutukset Ca2+ -virtoihin, LTP:hen, cAMP-tuotantoon ja muihin signalointikaskadeihin niiden signalointikyvyn määrittämiseksi. Nämä kokeet auttavat täyttämään joitakin nykyisen kirjallisuuden aukkoja AMYR:n ja sen osallisuuden osalta tautitiloissa (kuva 1).
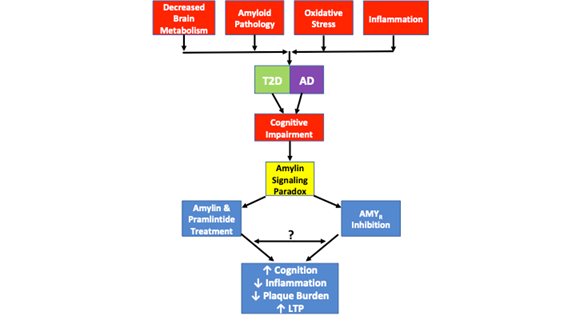
Kuvassa 1. on kuvattu T2D:ssä ja Alzheimerin taudissa havaittu amyliinin signalointiparadoksi ja patologiset yhtäläisyydet. Aivojen heikentynyt aineenvaihdunta, amyloidipatologia, oksidatiivinen stressi ja tulehdus ovat yhteisiä patologisia piirteitä, joita havaitaan molemmissa sairauksissa. Vaikka jokaisessa T2D- tai AD-tapauksessa ei ole kaikkia näitä patologisia piirteitä, kaikissa tapauksissa esiintyy kognitiivista heikentymistä. Amylinin signalointiparadoksi tulee kuvaan mukaan, sillä tutkimukset ovat osoittaneet, että sekä AMYR:n esto että AMYR-agonismi amylinin ja pramlintidihoidon kautta johtaa kognition paranemiseen, tulehduksen vähenemiseen, plakkitaakan vähenemiseen ja LTP:n lisääntymiseen. AMYR-agonismin ja AMYR-antagonismin hallitsemia signalointimekanismeja ei ole vielä täysin selvitetty. Vaikka amyliinin signalointi on perinteisesti liitetty cAMP- ja PKA-signalointiin, on epäselvää, aktivoivatko amyliini/pramlintidi myös muita kaskadeja. Lisäksi on epäselvää, viestivätkö AMYR-antagonistit, amyliini-oligomeerit tai Aβ AMYR:n kautta tai onko kaikkien näiden AMYR-ligandien välillä yhtäläisyyksiä tai ristikkäisvaikutuksia. Sellaisenaan useat tässä katsauksessa ehdotetut kokeet auttavat edelleen selvittämään AMYR:n todellisen luonteen.
Johtopäätökset
Tämänhetkinen epäjohdonmukaisuus amyliinisignaalin roolin suhteen aivoissa osoittaa, että amyliinin osallisuutta sekä Alzheimerin tautiin (AD) että T2D:hen on ehdottomasti selvitettävä lisää. T2D:ssä on todennäköistä, että taudin alkuvaiheessa amyliini tulvii aivoihin, muodostaa oligomeerejä, indusoi poikkeavaa signalointia natiivireseptorinsa kautta ja rekrytoi TRPV4:n indusoimaan patologisen Ca2+-tulvan, joka johtaa laajalle levinneeseen neuronaaliseen toimintahäiriöön, joka ilmenee OS:nä, kontrolloimattomana vesikulaarisena vapautumisena ja interneuronaalisena toimintahäiriönä, tulehduksina ja niistä johtuvana solukuolemana. Tämä mekanismi voi olla vastuussa alkuvaiheen siirtymisestä terveistä aivoista aivojen vanhenemiseen metabolisessa sairaudessa. Näin ollen AMYR:n tai TRVP4:n estäminen metabolisen sairauden tiettyinä ajankohtina ja diabeteksen alkuvaiheessa voi olla perusteltua oligomeerisen amyliinin tai Aβ:n toksisten vaikutusten estämiseksi. Vahvat todisteet viittaavat kuitenkin myös siihen, että amyliinin korvaaminen joko ihmisen amyliinilla tai pramlintidilla vähentää suurinta osaa AD:hen liittyvästä patologiasta ja parantaa samalla kognitiota AD:n jyrsijämalleissa. Näin ollen amyliinisignaalin korvaaminen amyliinillä tai pramlintidilla diabeteksen keski- ja myöhäisvaiheessa, kun amyliinisignaalin välittäminen on kadonnut aggregaation, oligomerisaation tai β-solukaton vuoksi, voi olla perusteltua. Tätä varten on myös tarpeen erottaa patologisten tapahtumien ajallinen esiintyminen aineenvaihduntaan liittyvässä aivojen vanhenemisessa ja terapeuttiset vaihtoehdot varhais-, väli- ja myöhäisvaiheen tautia varten. Näiden amyloidien suoran luonteen ja signalointikyvyn sekä tiettyjen ajallisten hoitojen terapeuttisen luonteen kriittinen analyysi ja testaus voivat auttaa kuromaan umpeen AMYR:n estohoitojen ja amyliinikorvaushoitojen välisen kuilun.
Rahoitus
Tämän artikkelin rahoitus saatiin National Institutes of Agingin apurahasta 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimerin tauti: Aβ, tau ja synaptinen toimintahäiriö. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurologia. 2013; 80: 1778-1783.
- Yhdistys As. 2016 Alzheimerin taudin faktat ja luvut. Alzheimerin & dementia. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimerin taudin lääkekehitysputki: vähän ehdokkaita, usein epäonnistumisia. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glukoositasot ja dementiariski. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta-analyysi ja mukautettu ennuste dementian esiintyvyydestä Yhdysvalloissa ja Kiinassa. Obesity. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Tyypin 2 diabetes Alzheimerin taudin riskitekijänä: tähän suhteeseen liittyvät sekoittajat, vuorovaikutukset ja neuropatologia. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurologia. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Lihavuus ja sen liitännäissairaudet. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF:n diabetesatlas. Bryssel: Kansainvälinen diabetesliitto. 2013.
- K Dash S. Kognitiivinen heikentyminen ja diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Dementia ja kognitiivinen heikkeneminen tyypin 2 diabeteksessa ja diabetesta edeltävissä vaiheissa: kohti kohdennettuja interventioita. The lancet Diabetes & endokrinologia. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalence of diabetes and prediabetes and their risk factors among Bangladeshi adults: a nationwide survey. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insuliinia hajottava entsyymi säätelee insuliinin, amyloidi β-proteiinin ja β-amyloidin esiasteproteiinin solunsisäisen domeenin tasoja in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insuliinin sitoutuminen aivokapillaareihin vähenee geneettisesti liikalihavilla, hyperinsulinemisilla Zucker-rotilla. Peptidit. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Insuliinitasot ovat alentuneet aivo-selkäydinnesteessä naisilla, joilla on prodominaalinen Alzheimerin tauti. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, ym. hAmyliinin moninkertainen kohde rotan primaarisissa hippokampuksen neuroneissa. Neurofarmakologia. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mekanismit, jotka yhdistävät lihavuuden insuliiniresistenssiin ja tyypin 2 diabetekseen. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. The American journal of pathology. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. NLRP3-inflammasomin aktivoituminen saarekeamyloidipolypeptidillä tarjoaa mekanismin lisääntyneelle IL-1β:lle tyypin 2 diabeteksessa. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimerin β-amyloidi, ihmisen saarekkeen amyliini ja prioniproteiinifragmentti aiheuttavat solunsisäisiä vapaan kalsiumin kohoamisia yhteisellä mekanismilla hypotalamuksen GnRH-neuronaalisessa solulinjassa. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo kylvö ja paikallisen amyloidoosin ristiinkylvö: molekulaarinen yhteys tyypin 2 diabeteksen ja Alzheimerin taudin välillä. The American journal of pathology. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylin-reseptori: yhteinen patofysiologinen kohde Alzheimerin taudissa ja diabetes mellituksessa. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ) -peptidi aktivoi suoraan amylin-3-reseptorin alatyypin laukaisemalla useita solunsisäisiä signaalireittejä. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintidi antagonisoi beeta-amyloidin (Aβ) ja ihmisen amyliinin aiheuttamaa Hippokampuksen pitkäaikaisen potentiaation depressiota. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloidin aiheuttama hippokampuksen pitkäaikaispotentiaation depressio välittyy amyliinireseptorin kautta. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Haiman peptidi amyliinin vatsansisäinen injektio vähentää tehokkaasti käyttäytymisen heikkenemistä ja aivojen amyloidipatologiaa Alzheimerin taudin hiirimalleissa. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylin-reseptoriligandit vähentävät Alzheimerin taudin patologista kaskadia. Neurofarmakologia. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. amyliinianalogin pramlintidin neuroprotektiiviset vaikutukset Alzheimerin taudin patogeneesiin ja kognitioon. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. β-kateniinin destabilisaatio presenilin-1:n mutaatioilla potensoi hermosoluapoptoosia. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin-hoito vähentää neuroinflammaatiota ja parantaa geeniekspression epänormaaleja kuvioita Alzheimerin taudin hiirimallin aivokuoressa. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintidiasetaatti-injektio tyypin 1 ja tyypin 2 diabetes mellituksen hoitoon. Clin Ther. 2007; 29: 535-562.
- Singh?Franco D, Perez A, Harrington C. Pramlintidiasetaatin vaikutus glykeemiseen kontrolliin ja painoon tyypin 2 diabetes mellitusta sairastavilla potilailla ja liikalihavilla potilailla, joilla ei ole diabetesta: systemaattinen katsaus ja meta-analyysi. Diabetes, Obesity and Metabolism. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. β-amyloidiproteiinin vaikutukset ihmisen neuroneihin ilmenevät amyliinireseptorin kautta. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyclic AC253, a novel amylin receptor antagonist, improves cognitive deficits in a mouse model of Alzheimer’s disease. Alzheimerin & dementia: Translational Research & Kliiniset interventiot. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Pramlintidin, rotan ja ihmisen amyliinin mutta ei Aβ1-42:n aktiivisuus ihmisen amyliinireseptoreissa. Endokrinologia. 2014; 155: 21-26.