Introduction
Sukulinjan de novo -mutaatiot (de novo mutations, DNM:t) ovat geneettisiä muutoksia yksilössä, jotka johtuvat vanhemmissa sukusoluissa tapahtuvasta mutaatioiden synnystä oogeneesin ja spermatogeneesin aikana. Tässä yhteydessä termiä ”de novo” ei pidä sekoittaa termiin ”uusi mutaatio”. Huolimatta siitä, että DNM:t kolmikon (isä, äiti ja lapsi) yhteydessä ovat uusia mutaatioita, ne voivat olla yleisiä, harvinaisia tai uusia variantteja yleisessä väestössä. Tietyn DNM:n esiintymistiheyden mittaamiseksi ja selittämiseksi on ensin arvioitava variantin vaikutus fenotyyppiin, koska uusia suotuisia ominaisuuksia voi kehittyä, kun syntyvät geneettiset mutaatiot tarjoavat tiettyä selviytymishyötyä (Front Line Genomics, 2017).
Ihmisillä, joilla on geneettisiä, ei-mendeliläisiä sairauksia, jotka esiintyvät satunnaisesti, DNM:t ovat yleensä uusia, luotettavampia ja haitallisempia kuin perinnölliset variantit, koska ne eivät ole voimakkaan luonnollisen valinnan alaisia (Crow, 2000; Front Line Genomics, 2017). Siksi DNM:n yksilössä aiheuttaman häiriön geneettisen syyn tunnistaminen voi olla kliinisestä näkökulmasta haastavaa, koska yhden fenotyypin taustalla voi olla pleiotropiaa ja geneettistä heterogeenisuutta (Eyre-Walker ja Keightley, 2007). Tämän vuoksi viime vuosikymmenen aikana on tehty huomattavia ponnisteluja eksomien sekvensoimiseksi yksilöiltä, joilla on epäselvää geneettistä etiologiaa omaavia sairauksia, kliinistä diagnostiikkaa varten. Kuitenkin jopa de novo -ehdokkaiden varianttien havaitsemisen jälkeen ei ole vielä riittävästi tietoa yleisistä ja harvinaisista varianteista, mikä estää selkeän johtopäätöksen tekemisen havaitun de novo -variantin patogeenisuudesta ja sen roolista sairaudessa (Acuna-Hidalgo ym., 2016). Tämä rajoitus saattaa selittyä sillä, että de novo -muunnokset ovat yleensä heterotsygoottisia ja voivat olla joko erittäin harvinaisia tai yleisiä. Erittäin harvinaisten de novo -varianttien tapauksessa variantin patogeenisuutta voi olla vaikea todistaa, koska potilaita, joilla on sama fenotyyppi ja de novo -variantti, ei ole enempää. Yleisten de novo -varianttien tapauksessa ei välttämättä tunneta tekijöitä, jotka määräävät variantin patogeenisuuden ilmenemismuodot, erityisesti jos joillakin väestöön kuuluvilla henkilöillä on kyseinen variantti, mutta heillä ei ole geneettistä sairautta. Riippumatta de novo -varianttien esiintymistiheydestä molemmat varianttien tyypit voidaan kuitenkin skaalata suhteellisen kelpoisuuden ja luonnollisen valinnan perusteella.
Sopeutuneisuus riippuu monista tekijöistä; siksi sen arvioimiseksi, onko DNM patogeeninen vai adaptiivinen, ja sen ymmärtämiseksi, miksi sitä esiintyy tietyllä frekvenssillä populaatiossa, on tutkittava variantti sopivissa olosuhteissa. Näitä ovat ympäristö, vanhempien ikä, genominen konteksti, epigenetiikka ja muut tekijät, koska ne kaikki vaikuttavat keskimääräisen suhteellisen kelpoisuuden arvoon, joka kasvaa monotonisesti, kun taas valinnan voimakkuus vähenee (Peck ja Waxman, 2018).
Tämän tutkimuksen päätavoitteena oli selvittää esiintyvien DNM-mutaatioiden esiintymisnopeus ja määritellä, miten nämä mutaatiot jakaantuvat yleisen liettualaisväestön eksomeissa. Tutkimme myös, vaikuttivatko näiden mutaatioiden esiintymistiheyteen niiden sekvenssien koostumus tai rakenneparametrit, joissa mutaatioita esiintyi, sekä muut tekijät, jotka voivat vaikuttaa näiden DNM:ien muodostumisen taustalla oleviin mekanismeihin. Lopuksi pyrimme selvittämään, syntyivätkö DNM:t toiminnallisiin alueisiin kohdistuvan voimakkaan luonnonvalintapaineen vuoksi. Vaikka DNM:ien jakautuminen ja voimakkuus ovat olleet monien tutkimusten aiheena, niitä ei ollut aiemmin tutkittu Liettuan väestössä.
Materiaalit ja menetelmät
Tässä tutkimuksessa analysoimme Liettuan väestöstä otettuja näytteitä, jotka saatiin LITGEN-hankkeesta (LITGEN, 2011). Aineisto koostui 49 kolmikosta, joissa oli yhteensä 144 eri yksilöä. Genominen DNA uutettiin laskimoverestä joko fenoli-kloroformiuuttomenetelmällä tai automatisoidulla DNA:n uuttamisalustalla TECAN Freedom EVO® (Tecan Schweiz AG, Sveitsi), joka perustuu paramagneettisten hiukkasten menetelmään. Eksomit sekvensoitiin SOLiD 5500 -sekvensointijärjestelmällä (75 bp:n lukemat). Sekvensointitiedot käsiteltiin ja valmisteltiin Lifescope-ohjelmistolla. Eksomit kartoitettiin ihmisen referenssigenomin build 19 mukaisesti. Sekvensoinnin keskimääräinen lukusyvyys oli 38,5. Lifescope-ohjelmalla tuotetut äidin, isän ja lapsen BAM-muotoiset tiedostot yhdistettiin SAMtools-ohjelmistolla kunkin kolmikon osalta.
De novo -mutaatiot tunnistettiin kahdella ohjelmalla: VarScan (Koboldt et al., 2012) ja VarSeqTM. Potentiaalinen variantti katsottiin DNM:ksi, jos se tunnistettiin jälkeläisissä, mutta sitä ei esiintynyt kummassakaan vanhemmassa samassa paikassa. Kaiken kaikkiaan VarScan havaitsi 1 752 ja VarSeqTM 4 756 DNM:ää. Väärien positiivisten de novo -kutsujen hylkäämiseksi, kun ei tiedetty, oliko kaikki kolmikon yksilöt tunnistettu oikein, käytettiin havaittujen DNM-laatuparametrien konservatiivisia suodattimia seuraavasti: (1) yksilön genotyypin laatu ≥50; (2) lukujen määrä kussakin kohdassa >20. SnpSift-ohjelmistoa käytettiin näiden suodattimien soveltamiseen VarScanin tuottamiin tietoihin. VarSeqTM -ohjelmiston tuottamat tiedot suodatettiin valitsemalla samat suodatusparametrit Trio Workflow -segmentissä. Lisäksi jäljelle jääneiden somaattisten (vain murto-osassa sekvensoiduista verisoluista esiintyvien) varianttien, joilla oli alhainen alleelitasapaino tai sekvensointiartefakteja, hylkäämiseksi DNM:t suodatettiin asettamalla kynnysarvo havaitulle lukemien osuudelle yksilöissä, joilla oli vaihtoehtoinen alleeli (alleelitasapaino) triolle (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Lisäksi kaikki mahdolliset tunnistetut ja suodatetut de novo -yksinukleotidivariantit tarkistettiin manuaalisesti Integrative Genomics Viewer -ohjelmalla (Robinson ym., 2011). Tunnistettujen DNM:ien suuren määrän vuoksi varianttien validointia varten Sanger-sekvensoinnilla valittiin satunnaisesti 51 de novo -yksinukleotidivarianttia. Sanger-sekvensointi suoritettiin ABI PRISM 3130xl Genetic Analyzer -laitteella. Kaikki VarScanilla (N = 95) ja VarSeqTM:llä (N = 84) tunnistetut suodatetut ja manuaalisesti tarkistetut DNM:t annotoitiin ANNOVAR-ohjelmalla (Butkiewicz ja Bush, 2016; Wang ym., 2010). Proteiinien vuorovaikutusten analysointiin käytettiin STRING-ohjelmistoa (Szklarczyk et al., 2017). Kuten eksomikartoituksessa, annotaatiot tehtiin käyttäen hg19-viite-ihmisen genomia.
Todennäköisyys sille, että kutsuttu positio oli DNM kolmikossa, laskettiin itsenäisesti jokaiselle kolmikolle. Kuten aiemmassa viitteessä (Besenbacher et al., 2015), de novo rate per position per generation (PPPG) laskettiin seuraavasti:
missä f on triojen lukumäärä ja N on niiden kutsuttavien paikkojen lukumäärä, jotka voidaan mahdollisesti tunnistaa de novo -paikoiksi jokaiselle triolle erikseen sekvensointisyvyydestä riippumatta. Tämä määrä vaihtelee triosta riippuen. ni on tunnistettujen DNM-kohteiden määrä triossa i. Todennäköisyys Pji (de novos ingle nucleotide) sille, että kutsuttu yksittäinen nukleotidipaikka j ja perhe i ovat mutaatioita, laskettiin seuraavasti:
Todennäköisyys Pji (de novo indel)kutsutulle indel-kohdalle j ja perheelle i olla mutatoitunut laskettiin seuraavasti:
missä C, M ja F tarkoittavat vastaavasti jälkeläistä, äitiä ja isää, ja Hetero, HomR ja HomA tarkoittavat vastaavasti heterotsygoottista, referenssihomotsygoottista ja homotsygoottista vaihtoehtoista alleelia. Todennäköisyys Pij (de novo) laskettiin suhteessa sekvensoinnin kattavuuteen. Suhde-estimaattien luottamusvälit laskettiin kuten binomisille osuuksille. DNM-asteen estimointiin ja muihin laskelmiin käytettiin R-pakettia (versio 3.4.3) (R Core Team, 2013).
Testaamaan hypoteesia, jonka mukaan DNM-asteen vaihtelut genomin eri alueilla voitaisiin selittää itse genomialueen luontaisilla ominaisuuksilla ja vanhempien iällä, suoritettiin lineaarinen regressioanalyysi, jota varten kunkin DNM:n ”toissijainen” annotaatio tehtiin ENCODE- (ENCODE Project Consortium, 2012) ja LITGEN-hankkeista (LITGEN, 2011) saatujen tietojen avulla. Ensin valittiin aikaisemman tutkimuksen (Besenbacher ym., 2015) mukaisesti (ENCODE Project Consortium, 2012) lymfoblastoidiset solulinjat (LCL ja GM12878), jotta voitiin kerätä tunnistettujen DNM:ien genomimaisemaa koskevia tietoja. Tietoja kerättiin:
(1) ilmentymisasteista (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) eri kudoksissa. Ekspression mukaan alueet, joilla oli DNM:iä, jaettiin positioihin, joilla oli spesifinen ja epäspesifinen ekspressio;
(2) DNaasi1-hypersensitiivisyyskohtien (DHS) mittaukset. DHS-status määritettiin 0:ksi, jos se oli DHS-piikin ulkopuolella, ja 1:ksi, jos se oli sen sisällä;
(3) CpG-saarekkeiden kontekstin mittaukset. Jos DNM oli CpG-saarten sisällä, aseman statukseksi annettiin 1; jos ulkopuolella – 0;
(4) kolme histonimerkkiä (H3K27ac, H3K4me1 ja H3K4me3) ENCODE-projektista. Jos DNM oli histonilla merkityssä paikassa, sille annettiin 1 ja jos ei – 0;
(5) GERPP++-konservointiarvot kerättiin ANNOVAR-annotaatiotyökalulla. Konservointiarvojen mukaan DNM:n sisältämät asemat jaettiin konservatiivisiin (GERP++-pisteet >12) ja ei-konservatiivisiin asemiin (GERP++-pisteet <12) (Davydov ym., 2010; ENCODE Project Consortium, 2012). LITGEN-hankkeen kyselylomaketietojen perusteella kerättiin tiedot vanhempien iästä. Kun parametrit oli kerätty kustakin kolmikosta, laskettiin kunkin parametrin sisältävien kantojen lukumäärä. Sitten suoritettiin korrelaatioanalyysi, jota seurasi DNM-asteen ja parametrien lineaarinen regressiomallinnus.
Tulokset
DNM-analyysin jälkeen kahdesta kolmikosta (nro 4 ja nro 21) tunnistettiin poikkeuksellisen suuri määrä DNM:iä: 113 ja 123 (VarScan- ja VarSeqTM-menetelmällä) ja 16 (VarScan). Nämä havainnot saivat meidät testaamaan biologista isyyttä, joka hylättiin kolmikon nro. 4 hylättiin ja vahvistettiin kolmikon nro 4 osalta. 21. Näin ollen kolmikon nro. 4 jätettiin tutkimuksen ulkopuolelle. Lopullisessa 48 kolmikon joukossa tunnistettiin VarScan-ohjelmistolla 95 DNM:ää 34 kolmikosta ja VarSeqTM-ohjelmistolla 84 DNM:ää 31 kolmikosta (kuva 1). VarScan-ohjelmistolla 18:ssa ja VarSeqTM-ohjelmistolla 15:ssä triossa ei havaittu yhtään DNM:ää. Kaikista DNM:istä, jotka tunnistettiin molemmilla ohjelmistoilla, vain 5,37 prosenttia DNM:istä vastasi toisiaan (kolme DNM:ää MEIS2-, PGK1- ja MT1B-geeneissä). Kullakin henkilöllä oli keskimäärin 1,9 (VarScan-ohjelmisto) ja 1,7 (VarSeqTM) DNM:ää.
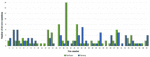
KUVIO 1. JOHDANTO. VarScan-ohjelmistolla (sininen) ja VarSeqTM-ohjelmistolla (vihreä) tunnistettujen yksittäisten de novo -nukleotidivarianttien vertailu.
Analyysi 95:stä VarScan-ohjelmistolla tunnistetusta DNM:stä osoitti, että 20 DNM:stä oli eksonisia, mukaan lukien kaksi stop-gain-DNM:ää, seitsemän synonyymistä DNM:ää ja 11 ei-synonyymistä DNM:ää. VarSeqTM-ohjelmalla tunnistetut 80 uutta mutaatiota olivat eksonisia, mukaan lukien 1 stop-gain DNM ja 78 ei-synonyymistä DNM:ää (kuva 2). Suurin osa VarScanilla tunnistetuista DNM:istä oli kromosomeissa 1, 2, 4 ja 5, kun taas VarSeqTM tunnisti DNM:iä pääasiassa kromosomeissa 2, 6, 7 ja 11. Tunnistettujen DNM:ien määrä ei korreloinut geenien tiheyden kanssa kromosomeissa (R = 0,09, p-arvo = 0,65 VarScanilla ja R = 6,73, p-arvo = 0,51 VarSeqTM:llä) eikä kromosomien koon kanssa (kuva 3). Molempien ohjelmien mukaan siirtymien ja transversioiden suhdeluvut olivat hyvin samanlaiset: 1,44 ja 1,47 (kuva 4). Siirtymien rakenteissa havaittiin kuitenkin eroja. Erityisesti VarScanilla tunnistetuissa DNM:ssä oli enemmän G/T- ja A/C-muutoksia, kun taas VarSeqTM:llä tunnistetuissa DNM:ssä oli enemmän A/T- ja G/C-muutoksia.
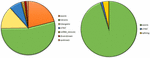
KUVIO 2. DNM:t. VarScanin (vasemmalla) ja VarSeqTM:n (oikealla) tuottamien de novo -mutaatioiden (DNM) koostumus.
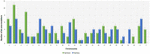
KUVIO 3. De novo -mutaatioiden (DNM) koostumus. De novo -varianttien lukumäärän jakautuminen kromosomeittain VarScanin ja VarSeqTM:n tuottaman datan mukaan. Vihreät palkit kuvaavat VarScan-ohjelmistolla tunnistettuja DNM:iä, siniset – VarSeqTM-ohjelmistolla tunnistettuja DNM:iä. Virhepalkit edustavat kunkin kromosomin DNM-keskiarvojen keskivirhettä.
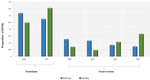
KUVIO 4. DNM-keskiarvojen keskivirhe. Siirtymien taustalla olevia molekyylitapahtumia esiintyy useammin kuin transversioihin johtavia tapahtumia, mikä johtaa siihen, että koko eksomissa on ∼1,5-kertaisesti enemmän siirtymiä kuin transversioita. VarScan- (vihreä) ja VarSeqTM-ohjelmistolla (sininen) tunnistetut siirtymä- ja transversiotapahtumat. Virhepalkit edustavat DNM-keskiarvojen keskivirhettä.
Yksittäisten nukleotidimutaatioiden de novo -mutaatioiden laskennallinen määrä oli 2,4 × 10-8 PPPG (95 %:n luottamusväli : 1,96 × 10-8-2,99 × 10-8) VarSeqTM -ohjelman mukaan ja 2.74 × 10-8 per nukleotidi per sukupolvi (95 %:n CI: 2,24 × 10-8-3,35 × 10-8) VarScan-algoritmin mukaan.
Kolme de novo -indeliä kolmessa triossa tunnistettiin VarScan-algoritmilla kromosomeissa 6 ja 11. De novo -indelien laskennallinen osuus genomissa oli 1,77 × 10-8 (95 % CI: 6,03 × 10-9-5,2 × 10-8) PPPG. Huomionarvoista on, että kaikki de novo -indelit olivat ”palautuvia”, eli vanhemmilla oli uusia variantteja genomissa, ja heidän lapsillaan oli de novo -variantteja, jotka perustuivat referenssigenomiin, jonka sekvensointisyvyyden keskiarvo oli 37,5 ja genotyypin laadun keskiarvo 50. Näitä kolmea DNM:ää ei kuitenkaan valittu validointiin Sangerin sekvensointimenetelmällä, joten de novo -indelien yliarvioinnin todennäköisyys säilyy kuitenkin. De novo -indelit olivat C/T ja A/G yksittäisten nukleotidien yhteydessä.
Lineaarinen regressiomallinnus osoitti, että DNAse 1 -hypersensitiivisyyspaikat, CpG-saarekkeiden konteksti, GERPP++ -säilyttävyysarvot ja ekspressiotasot selittivät ∼68-93 % DNM-asteista (taulukko 1). Epigeneettiset markkerit tai isän ikä eivät korreloineet merkittävästi DNM-asteen kanssa. Mallit laadittiin vain VarScanista saatujen tietojen perusteella, koska VarSeqTM:stä saatujen tietojen ja itse genomialueen luontaisten ominaisuuksien välillä ei ollut korrelaatiota.

TAULUKKO 1. JOHDANTO. DNAaseI-hypersensitiivisyyskohtien, CpG-saarekkeiden kontekstin, GERPP++-konservaatioarvojen ja ekspressiotason vaikutuksen lineaarinen regressio DNM:ien määrään.
DNM:ien funktionaalinen ennustaminen
Ja sen arvioimiseksi, mitkä missense-mutaatiot olivat haitallisia ja muuttivat vaikutuksen kohteena olleen valkuaisaineen funktiota tyypinomaisesti, analysoitiin DNM:ien aikaansaamien vaurioiden ennustettuja kategorisia pistemääriä. Seuraavat 10 arvoa otettiin huomioon: polyphen HDIV ja HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, Fathmm-MKL-koodaus ja GERP++. Ennustettujen pisteiden perusteella valittiin neljä VarScanin tunnistamaa DNM:ää, joilla oli kuusi tai useampia vahingollisia tai todennäköisesti vahingollisia ennusteita. Nämä stop-gain DNM:t olivat MEIS2- ja ULK4-geeneissä, kun taas ei-synonyymiset DNM:t olivat MT1B- ja PGK1-geeneissä. Näiden geenien koodaamat proteiinit ovat tärkeitä hermosolujen kasvulle, endosytoosille ja suojautumiselle raskasmetallien kielteisiltä vaikutuksilta. Nämä proteiinit osallistuvat kasvaimen verisuonia estävän angiostatiinin vapautumiseen ja erilaisiin signaalireitteihin. Näiden geenien koodaamien proteiinien välillä ei ollut yhteyksiä (kuva 5).
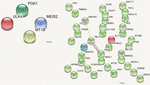
KUVA 5. Näiden geenien koodaamien proteiinien väliset yhteydet. Proteiini-proteiini-interaktiot (Szklarczyk ym., 2017) DNM:iä sisältävissä geeneissä. VarScanilla tunnistetut DNM:t proteiineja koodaavissa geeneissä ovat vasemmalla, VarSeqTM -ohjelmalla tunnistetut DNM:t oikealla. Värilliset viivat viittaavat proteiinien väliseen yhteyteen.
VarSeqTM:n tunnistamia de novo -mutaatioita analysoitiin tarkemmin, jos vähintään puolet ennustetyökaluista ennusti niiden olevan vahingollisia tai todennäköisesti vahingollisia. Pistemutaatioita oli 35 (ks. ??) geeneissä, jotka koodaavat proteiineja, jotka olivat tärkeitä kromatiinin uudelleenmuotoilussa, sytoskeletin säätelyssä, solujen kasvussa ja elinkelpoisuudessa, sytoplasmisissa signaalireiteissä ja hajun havaitsemisen laukaisevien hermovasteiden käynnistymisessä.
DNM-vaurioituneiden geenien koodaamista proteiineista vain CLPTM1, ZNF547 ja DMXL1 olivat jollakin tavalla yhteydessä toisiinsa (kuva 5).
Keskustelu
Tässä tutkimuksessa teimme kattavan analyysin DNM:ien jakaantumisesta liettualaispopulaatiossa eksomin eri alueille. SOLiD 5500 -sekvensointiteknologiaa käyttäen havaittiin VarScan- ja VarSeqTM-algoritmeilla yhteensä 95 DNM:ää 34 triossa ja 84 DNM:ää 31 triossa. Ensinnäkin haluamme huomauttaa, että valitsimme VarScan-algoritmin DNM:ien kutsumiseen, koska (Warden ym., 2014) mukaan tämä algoritmi tuottaa luettelon varianteista, joilla on korkea yhdenmukaisuus (>97 %) GATK UnifiedGenotyperin ja HaplotypeCallerin kutsumien korkealaatuisten varianttien kanssa. VarSeqTM-ohjelmisto valittiin, koska se on laajalti käytetty työkalu varianttien analysointiin sekä tutkimuksissa että kliinisissä analyyseissä. Huolimatta siitä, että molemmat algoritmit on suunniteltu etsimään jälkeläisen eksomista DNM:iä, joita ei ollut kummassakaan vanhemmassa, näiden kahden ohjelmiston välinen yksimielisyys DNM-analyysissä oli vain 5,37 prosenttia. VarScan-algoritmilla oli suurempi herkkyys (5,42 %) DNM:ien havaitsemisessa ennen suodatusta kuin VarSeqTM-algoritmilla (1,77 %), joten epäilimme, että yksikään työkalu ei onnistunut kutsumaan mutaatioita korkean herkkyyden vuoksi, johon liittyi aina alhainen spesifisyys. Siksi ehdotamme, että tuloksia voitaisiin parantaa huomattavasti yhdistämällä eri työkalujen tuotoksia (Sandmann ym., 2017).
Tuotetun datan perusteella arvioitu yhden nukleotidin DNM-taajuus oli 2,4 × 10-8 ja 2,74 × 10-8 välillä ja de novo indeleiden osuus oli 1,77 × 10-8 PPPG, riippuen käytetystä algoritmista. Laskemamme DNM-aste oli korkeampi kuin aiemmissa tutkimuksissa (Kong ym., 2010, 2012; Neale ym., 2012; Szamecz ym., 2014; Besenbacher ym., 2015; Francioli ym., 2015) raportoitu DNM-aste, jossa se vaihteli välillä 1,2 × 10-8 ja 1,5 × 10-8 PPPG. Tutkimuksemme korkeampi DNM-aste oli kohtuullinen, koska tutkimuksemme perustui eksomitietoihin. Lisäksi eksomeissa on huomattavasti korkeampi (30 %) mutaatioprosentti kuin kokonaisissa genomeissa, koska koko genomin emäsparikoostumus on erilainen kuin eksomeissa. Eksomien keskimääräinen GC-pitoisuus on noin 50 %, kun taas koko genomin GC-pitoisuus on noin 40 % (Neale et al., 2012). Metyloituneet CpG:t edustavat ihmisillä erittäin mutatoituvia sekvenssejä sytosiiniemästen spontaanin deaminoinnin vuoksi (Neale et al., 2012). Vertailevan genomiikan tutkimusten mukaan CpG-rikkaiden alueiden kohonneiden mutaatiomäärien uskotaan kehittyneen nisäkkäiden säteilyn aikaan (Francioli ym., 2015). Lajien erilaistumisen aikana CpG-rikkaat eksoniset alueet kokivat lisääntyneet mutaatioluvut verrattuna ei-koodaavan DNA:n mutaatiolukuihin ja muuttuivat ei-koodaaviksi alueiksi. Siksi sitten CpG-pitoisuuden vaikutus vähenee ajan myötä, keskimääräinen mutaationopeus laskee, kunnes se saavuttaa ympäröivän ei-koodaavan DNA:n tasolla (Subramanian ja Kumar, 2003). Kuitenkin siinä missä neutraalisti kehittyvillä genomin alueilla olevilla sekvensseillä on ollut riittävästi aikaa tasapainottua dinukleotidikontekstien suhteen, puhdistava valinta on ylläpitänyt hypermutaatioita aiheuttavia CpG:tä toiminnallisilla alueilla (Subramanian ja Kumar, 2003; Schmidt ym., 2008; Francioli ym., 2015). Siksi, koska havaitsimme korkeamman DNM-asteen kuin muissa tutkimuksissa raportoitu, spekuloimme, että se saattaa ainakin osittain johtua paikallisesta sekvenssikontekstista ja/tai mahdollisesta luonnollisesta valintapaineesta eksomissa. Näin ollen käytettiin lineaarista regressiomallia, ja havaitsimme, että DNAse 1 -hypersensitiivisyys, CpG-saarekkeiden konteksti, GERPP++-konservaatioarvot ja ekspressiotaso selittivät ∼68-93 % DNM-asteesta. Nämä havainnot osoittivat, että DNM:t eksomissa muodostuivat DNA-sekvenssien säilymisestä riippumatta. DNM-aste oli kuitenkin korkeampi geeneissä, joiden tuotteet olivat epäspesifisiä, ja transkriptiivisesti aktiivisilla promoottorin kaltaisilla alueilla.
Kontrastina muiden tutkimusten tuloksiin (Wong ym., 2016; Sandmann ym., 2017) havaitsimme, että isän ikä ei korreloinut DNM-asteen kanssa. Nämä havainnot voivat selittyä sillä, että aineisto koostui trioista, joiden vanhemmat olivat iältään samankaltaisia, ja että koko genomista analysoitiin vain pieni osa (∼1,5 %). Näiden parametrien perusteella jokaisella henkilöllä oli keskimäärin vain 1,9 (VarScan) tai 1,7 (VarSeqTM) DNM:ää verrattuna 40-82:een koko genomissa (Crow, 2000; Branciamore ym., 2010; Kong ym., 2012; Neale ym., 2012; Besenbacher ym., 2015; Francioli ym., 2015; Wong ym, 2016), kun taas koodaavassa sekvenssissä olevien de novo -indelien määrä oli samankaltainen kuin (Front Line Genomics, 2017).
Annotaatioiden laajan funktionaalisen analyysin tulokset paljastivat, että kaikista tunnistetuista DNM:istä 4 (VarScan) ja 35 (VarSeqTM) varianttia olivat todennäköisesti patogeenisiä DNM:iä. Ero patogeenisten DNM:ien määrässä saattaa selittyä sillä, että DNM:ien tunnistamiseen käytetystä algoritmista riippuen DNM:ien osuus koodaavissa sekvensseissä vaihteli merkittävästi. Esimerkiksi VarScan-ohjelmistolla tunnistetuista DNM:istä 21,05 prosenttia oli eksonisia, kun taas VarSeqTM-ohjelmistolla tunnistetuista DNM:istä 95,24 prosenttia oli eksonisia. Nämä patogeeniset DNM:t olivat geeneissä, jotka koodasivat proteiineja, jotka ovat välttämättömiä kromatiinin mallintamisessa, sytoskeletin säätelyssä, solun kasvun ja elinvoimaisuuden moduloinnissa, sytoplasman signaalireittien toiminnassa ja hermovasteen käynnistämisessä. Vaikka näitä DNM:iä pidettiin patogeenisina, kaikki kyselyyn osallistuneet henkilöt pitivät itseään geneettisesti ”terveinä”. Näin ollen tämä tulos osoitti, että huolimatta DNM:ien oletetusta patogeenisyydestä genomit, joissa DNM:t sijaitsivat, ilmeisesti sietivät tällaisia muutoksia, joten taudin ilmenemismuodot eivät useinkaan olleet voimakkaita. Szameczin ym. (2014) mukaan mitä useammin DNM:t esiintyvät konservoiduissa geneettisissä paikoissa, sitä voimakkaampia ovat luonnollisen valinnan vaikutukset geneettisiin muutoksiin kompensoivien genomin suojamekanismien kautta. Varianttien haitallisia vaikutuksia voidaan lieventää neljällä tavalla. Jotkin geenit voivat sietää proteiinien typistettyjä variantteja, koska niiden toiminnalliset vaikutukset peittyvät epätäydellisen ilmentymisen, kompensoivien varianttien tai typistyksen vähäisen toiminnallisen merkityksen vuoksi (Bartha ym., 2015). Sitä vastoin ei-synonyymisiin DNM:iin liittyvät geenimuutokset kompensoituvat hyödyllisen mutaatiokertymän mekanismin kautta koko genomissa (Szamecz ym., 2014). Se viittaa siihen, että näissä tapauksissa patogeeniset mutaatiot eivät ole niin haitallisia, että ne vähentäisivät keskimääräistä kuntoa, ja siksi ne säilyvät pidempään monissa sukupolvissa luonnollisen valinnan muokkaamina.
Yhteenvetona voidaan todeta, että analyysimme DNM:ien jakautumisesta ja niiden geneettisestä ja epigeneettisestä kontekstista antoi tietoa Liettuan genomin geneettisestä vaihtelusta. Näiden havaintojen perusteella lisätutkimukset geneettisiä sairauksia sairastavissa potilasryhmissä voivat helpottaa kykyämme erottaa tietyt patogeeniset DNM:t siedetyistä tausta-DNM:istä ja tunnistaa luotettavia aiheuttavia DNM:iä. Tämän tutkimuksen tärkein rajoitus oli kuitenkin se, että emme tutkineet ei-koodaavien ja säätelygeenialueiden vaihtelua. Nämä tiedot voisivat osaltaan auttaa selvittämään DNM:ien muodostumisen mahdollisia mekanismeja, jotka eivät ole vielä riittävän selkeitä.
Accession Codes
Sequence data has been deposited at the European Nucleotide Archive (ENA), under accession PRJEB25864 (ERP107829).
Ethics Statement
This study was carried carried out in accordance with the recommendations of permission, Vilnan alueellinen biolääketieteellistä tutkimusta käsittelevä eettinen komitea. Tutkimussuunnitelma hyväksyttiin Vilnan alueellisessa biolääketieteellisen tutkimuksen eettisessä komiteassa. Kaikki koehenkilöt antoivat kirjallisen tietoon perustuvan suostumuksen Helsingin julistuksen mukaisesti.
Author Contributions
LP suoritti tietojen analysoinnin ja laati käsikirjoituksen. AJ laski de novo -mutaatioiden määrän. Trioeksomien sekvensoinnin suorittivat LA ja IK. VK oli päätutkija.
Rahoitus
Tätä tutkimusta tuki Euroopan sosiaalirahasto Global Grant -toimenpiteen puitteissa. LITGEN-hanke nro. VP1-3.1-ŠMM-07-K-01-013.
Conflict of Interest Statement
Tekijät ilmoittavat, että tutkimus suoritettiin ilman kaupallisia tai taloudellisia suhteita, jotka voitaisiin tulkita mahdolliseksi eturistiriidaksi.
Supplementary Material
Tämän artikkelin lisäaineisto löytyy verkosta osoitteesta: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., ja Hoischen, A. (2016). Uutta tietoa de novo -mutaatioiden synnystä ja roolista terveydessä ja sairaudessa. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). Heterotsygoottisten proteiineja typistävien varianttien ominaisuudet ihmisen genomissa. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., ym. et al. (2015). Uudenlainen variaatio ja de novo -mutaatiomäärät populaation laajuisissa de novo kootuissa tanskalaisissa trioissa. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). CpG-saarekkeet ja CpG:n pro-epigeneettinen valinta HOX:n ja muiden transkriptiotekijöiden proteiinia koodaavissa eksoneissa. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). Genomisen variaation in silico funktionaalinen annotaatio. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Ihmisen spontaanin mutaation alkuperä, mallit ja vaikutukset. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Suuren osan ihmisen genomista tunnistaminen valikoivan rajoituksen alaiseksi GERP++:n avulla. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Ihmisen genomin DNA-elementtien integroitu tietosanakirja. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., ja Keightley, P. D. (2007). Uusien mutaatioiden kuntovaikutusten jakautuminen. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). De novo -mutaatioiden genominlaajuiset mallit ja ominaisuudet ihmisillä. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics -lehden numero 14 – ASHG. Lontoo: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Geneettiset vaikutukset geeniekspressioon eri ihmiskudoksissa. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: somaattisten mutaatioiden ja kopionumeromuutosten löytäminen syövässä eksomisekvensoinnin avulla. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). De novo -mutaatioiden määrä ja isän iän merkitys tautiriskiin. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., ym. et al. (2010). Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ’t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Transkriptomin ja genomin sekvensointi paljastaa toiminnallista vaihtelua ihmisillä. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Saatavilla osoitteessa: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Eksonisten de novo -mutaatioiden mallit ja määrät autismin kirjon häiriöissä. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Mitä on sopeutuminen ja miten sitä pitäisi mitata? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). A Language and Environment for Statistical Computing. Wien: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S. ja Sunyaev, S. (2008). Hypermutable non-synonymous sites are under stronger negative selection. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Neutraaleja substituutioita tapahtuu nopeammin eksoneissa kuin ei-koodaavassa DNA:ssa kädellisten genomissa. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). Kompensoivan evoluution genominen maisema Be. The genomic landscape of compensatory evolution. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). STRING-tietokanta vuonna 2017: Laadunvalvotut proteiini-proteiini-assosiaatioverkot, jotka ovat laajasti saatavilla. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Kahden suositun varianttien kutsumispaketin yksityiskohtainen vertailu eksomi- ja kohdennettuja eksonitutkimuksia varten. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Uusia havaintoja äidin iän vaikutuksesta ituradan de novo -mutaatioihin. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar