John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Des preuves croissantes mettent en évidence la relation intime entre le diabète de type II (T2D) et la maladie d’Alzheimer (AD). Fait important, ces deux maladies partagent un certain nombre de similitudes pathologiques, notamment l’accumulation d’amyloïdes, le stress oxydatif, l’inflammation et la mort cellulaire. À ce jour, les thérapies médicamenteuses pour la MA et le DT2 font défaut et il existe un besoin crucial de découvrir et de développer de nouvelles thérapies pour ces maladies. Un certain nombre d’études menées chez l’homme et chez les rongeurs ont démontré que la supplémentation en hormones métaboliques est très utile pour améliorer la fonction cognitive et la santé métabolique globale dans le DT2 et la MA. L’amyline, une hormone pancréatique, est apparue comme un élément crucial de l’étiologie du DT2 et de la MA, bien que le rôle exact de l’amyline dans ces maladies ne soit pas encore bien compris. Ici, nous passons en revue de manière critique la littérature actuelle qui utilise l’amyline humaine ou son analogue synthétique, le pramlintide, ainsi que les antagonistes des récepteurs de l’amyline pour le traitement de la MA.
Introduction
La maladie d’Alzheimer (MA) est une maladie neurodégénérative progressive et débilitante caractérisée par l’accumulation de plaques de bêta-amyloïde (Aβ) et d’enchevêtrements neurofibrillaires composés de tau1 hyperphosphorylé. L’accumulation de ces peptides pathologiques contribue aux déficits des fonctions exécutives telles que l’apprentissage et la mémoire, l’humeur, l’affect, etc. et représente un fardeau substantiel pour le patient et les soignants. L’incidence de la MA augmente à un rythme alarmant aux États-Unis, où l’on estime que 5,5 millions d’Américains vivent avec la MA en 2017 et que ce nombre devrait tripler d’ici 20502. En outre, le coût de la prise en charge et du traitement des patients atteints de la MA dépasse actuellement 200 milliards de dollars par an et devrait continuer à augmenter3. Bien que la MA soit clairement un problème monumental aux États-Unis et ailleurs, les options de traitement restent très limitées4. De nombreux essais de médicaments ont été menés avec un large éventail d’approches ciblées, mais il n’y a actuellement que six médicaments approuvés par la FDA pour la MA et il ne s’agit que de traitements symptomatiques5, 6. À ce jour, la majorité des agents pharmacologiques développés ont ciblé spécifiquement la pathologie Aβ ou tau, mais aucun n’a réussi à éliminer ou à prévenir la pathologie4. Il existe donc un besoin fondamental de développer des traitements thérapeutiques et préventifs viables pour la MA.
La MA liée à l’âge (sporadique) est une maladie multifactorielle compliquée, ayant de nombreuses influences génétiques et environnementales. L’environnement et le mode de vie sont fortement impliqués dans le développement de la MA sporadique ; des facteurs tels que l’alimentation7-9, l’obésité8-10, le syndrome métabolique7, le diabète de type II (DT2)9, 11 et les maladies cardiovasculaires12 ont tous été impliqués dans la causalité de la MA. Il est essentiel de noter que les taux d’obésité et de diabète augmentent rapidement, parallèlement à la MA12, 13. Bien que la relation entre l’obésité et la MA ne soit pas très claire, il est prouvé que l’obésité au milieu de la vie joue un rôle dans le développement de la MA10. Plus important encore, l’obésité s’accompagne généralement d’un certain nombre d’autres maladies, notamment les maladies cardiovasculaires, l’hypertension, la dyslipidémie, le DT2, les accidents vasculaires cérébraux, etc.14. L’incidence du DT2 augmente rapidement, les CDC estimant qu’environ 30,3 millions de personnes (1 adulte sur 10) sont diabétiques aux États-Unis et qu’un nombre stupéfiant de 84,1 millions (1 adulte sur 3) sont prédiabétiques, la plupart d’entre elles ignorant leur état. En outre, en raison d’une diminution à grande échelle de l’activité physique qui s’accompagne d’une augmentation simultanée de l’apport alimentaire et d’une mauvaise alimentation, les taux d’obésité, de DT2, de syndrome métabolique et de maladies cardiovasculaires ne peuvent qu’augmenter pour atteindre un nombre de cas de DT2 estimé à 600 millions dans le monde d’ici 203515.
L’ensemble des preuves impliquant la fonction et la maladie métaboliques dans le processus de déclin cognitif et de vieillissement est substantiel16, 17. Par exemple, environ 70 % des personnes diagnostiquées avec un DT2 font état de troubles cognitifs et un nombre important de patients atteints de DT2 développent plus tard une démence16, 18-21. Les personnes chez qui on a diagnostiqué un DT2 depuis au moins cinq ans ont un risque significativement plus élevé de développer une MA que celles qui souffrent d’un DT2 depuis moins de cinq ans17. L’ensemble de ces données suggère que la prévalence croissante du DT2 dans la population pourrait contribuer à l’augmentation des taux de MA.
Le DT2 se caractérise initialement par une glycémie et une insuline élevées, ce qui entraîne une hyperinsulinémie ; il est important de noter que l’amyline, une petite hormone métabolique produite par les cellules des îlots β du pancréas, est co-emballée et co-sécrétée avec l’insuline et est donc surproduite dans le DT222. Il est important de noter qu’un certain nombre de caractéristiques pathologiques sont présentes à la fois dans le DT2 et la MA : 1) diminution du métabolisme cérébral et résistance aux hormones métaboliques ; 2) pathologie amyloïde ; 3) stress oxydatif (SO) et inflammation. L’hyperinsulinémie et l’hyperamylinémie chroniques entraînent un certain nombre de problèmes physiologiques : l’hyperinsulinémie chronique entraîne une insulinorésistance systémique22, une altération du transport de l’insuline à travers la barrière hémato-encéphalique (BHE)23, 24, et donc une diminution de la signalisation de l’insuline dans le cerveau25. La perte de la signalisation de l’insuline dans le cerveau est associée à un certain nombre de caractéristiques pathologiques liées à la MA, notamment une augmentation de la production d’Aβ, de la phosphorylation de tau et de la neuroinflammation.
En outre, l’amyline partage des caractéristiques pathologiques similaires à celles de l’Aβ à des concentrations élevées26 et pourrait constituer une voie commune entre les deux maladies. Par exemple, des fibrilles d’amyline ont été trouvées dans le pancréas de 95 % des patients atteints de DT227-29 et provoquent un certain nombre de perturbations physiologiques, notamment un influx aberrant de Ca2+, une augmentation de la sécrétion de cytokines pro-inflammatoires30,31 et, finalement, la perte de cellules β-îlots32. En outre, l’amyline traverse facilement la BHE et forme des fibrilles d’amyline ainsi que des plaques mixtes avec de l’Aβ dans le cerveau et pourrait être responsable de la pathologie de type MA et de l’ensemencement en Aβ dans le DT233-35. L’amyline est connue pour affecter la potentialisation à long terme (LTP) dans l’hippocampe et pourrait avoir une influence innée sur la fonction cognitive dans le cerveau36-39. Cependant, on ne sait toujours pas si l’amyline est une insulte toxique dans ces maladies ou si sa perte fonctionnelle par agrégation ou par perte de cellules β au stade tardif dans le DT2 contribue au développement d’une MA.
La dichotomie de la signalisation de l’amyline
Il y a encore beaucoup de débats sur l’implication du récepteur de l’amyline (AMYR) et de la signalisation de l’amyline dans la progression de la maladie et l’étiologie du DT2 et de la MA. Le corpus de recherche visant à discerner cette relation s’élargit rapidement. Toutes les recherches pertinentes ont démontré de manière cohérente que la modulation de la signalisation de l’amyline affecte la pathologie liée à la MA. La nature de cette relation n’a toutefois pas encore été concrètement élucidée. Plusieurs groupes ont produit des données convaincantes suggérant que la signalisation de l’amyline est bénéfique pour prévenir la pathologie liée à la MA et les déficits cognitifs, à la fois in vivo et in vitro40-44. Il est important de noter que le pramlintide, une forme recombinante non agrégative de l’amyline, utilisé en conjonction avec des thérapies à l’insuline pour traiter le diabète et améliorer le contrôle glycémique, réduire le poids corporel et diminuer les marqueurs sériques de l’OS45-47, est également prometteur en tant que thérapie de la MA. Cependant, à ce jour, aucun essai clinique n’a visé à utiliser l’amyline ou le pramlintide comme agent thérapeutique dans le traitement de la démence. Les études sur les rongeurs ont clairement démontré que le traitement chronique par l’amyline ou le pramlintide humain présente un fort avantage thérapeutique en réduisant la pathologie liée à la MA ; la supplémentation en amyline/pramlintide réduit les niveaux d’Aβ soluble, la charge de la plaque, la phosphorylation de la protéine tau, la neuroinflammation et l’OS tout en améliorant la cognition40-42,44. Les données ci-dessus suggèrent qu’une perte de la signalisation innée de l’amyline dans le SNC en raison de l’agrégation donne lieu à un risque accru de développement de la MA et est couverte plus en détail dans Grizzanti et al. 201848.
En revanche, des études montrent également que l’amyline humaine et l’Aβ ont des effets toxiques similaires et que ces effets toxiques peuvent être atténués en utilisant l’antagoniste AMYR36-39,49. Par exemple, les données montrent qu’un traitement in vivo par des antagonistes de l’AMYR produit des avantages physiologiques très similaires à ceux d’un traitement par l’amyline ou le pramlintide. Le traitement de souris TgCRND8 AD avec AC253, un antagoniste AMYR, ou son homologue cyclique cAC253 réduit la neuroinflammation, les niveaux d’Aβ soluble et la charge de la plaque tout en améliorant la cognition50. De même, des études in vitro/ex-vivo montrent que de faibles doses d’amyline humaine ou d’Aβ provoquent des perturbations de la LTP et que ces déficits sont bloqués par l’AC253 ou le pramlintide38,39, et que des doses plus élevées d’amyline humaine/oligomères d’amyline sont associées à un influx incontrôlé de Ca2+, qui est fortement lié à la mort cellulaire26,32. Ensemble, ces données soutiennent une fonction toxique des oligomères d’amyline et donc un mécanisme thérapeutique potentiel pour le blocage de l’AMYR. En revanche, d’autres ont montré que les effets bénéfiques de l’amyline peuvent être bloqués par l’AC25341. Ainsi, le potentiel thérapeutique du traitement ou de l’inhibition de l’amyline reste flou et souligne la nature complexe et dichotomique des amyloïdes dans le cerveau et la périphérie.
Piecing Together the Puzzle
Il y a un certain nombre de trous dans la littérature actuelle qui doivent être comblés pour donner une image plus complète de l’histoire de l’amyline : 1) la nature du système inné de l’amyline et la signalisation de l’amyline dans le cerveau 2) les capacités de signalisation de l’Aβ et du pramlintide par les trois principaux AMYR et récepteurs apparentés 3) les mécanismes thérapeutiques par lesquels l’inhibition de l’amyline/pramlintide ou des AMYR sont médiés. Tout d’abord, de nouvelles données intéressantes démontrent que l’AMYR n’est pas seulement impliqué dans la signalisation, mais aussi dans le transport du ligand à travers la BHE. L’AMYR est un récepteur hétérodimérique qui se compose d’un récepteur de la calcitonine et d’une protéine modifiant l’activité du récepteur (1-3)51. À cette fin, un knockdown global de 50 % du récepteur de la calcitonine (un composant clé du RCAA) a réduit de manière significative la quantité d’AC253 trouvée dans le cerveau50, ce qui indique que le RCAA situé dans la BHE est impliqué dans le transport de ces ligands dans le cerveau et peut également être impliqué dans la circulation de l’amyline et du pramlintide dans/hors du cerveau. L’existence de ces mécanismes de transport dans la BHE suggère que l’amyline a probablement une fonction physiologique innée dans le cerveau, car son transport dans le cerveau est étroitement contrôlé. Cependant, on ne sait toujours pas comment la signalisation de l’amyline ou son absence conduit aux caractéristiques pathologiques de la MA et si la RVAA est le véhicule par lequel Aβ transmet ses effets toxiques.
Puis, des preuves contradictoires existent en ce qui concerne la relation entre Aβ et la RVAA. Bien que plusieurs études démontrent clairement que l’amyline humaine et l’Aβ ont des effets similaires sur la LTP dans le SNC et que l’utilisation d’inhibiteurs de l’AMYR améliore ces effets délétères36-39, d’autres preuves suggèrent que l’Aβ (1-42) est incapable de se signaler par l’intermédiaire de l’AMYR pour évoquer toute sorte de réponse AMPc à une grande variété de concentrations52. Il est possible que l’Aβ active différentes cascades de signalisation par le biais d’une interaction avec l’AMYR ou qu’il agisse simplement comme un inhibiteur compétitif inerte, mais cela reste à démontrer.
En outre, une étude distincte a démontré que l’amyline oligomère médiait ses effets toxiques directement par l’AMYR et indirectement par TRPV4, un canal cationique non sélectif26. De faibles concentrations d’amyline humaine provoquent une réponse Ca2+ qui est médiée par son récepteur natif. Cependant, à des concentrations plus élevées, l’amyline humaine forme des oligomères et active une signalisation aberrante qui entraîne l’activation des canaux TRVP4 et permet un afflux incontrôlé de cations, en particulier de Ca2+. Le blocage pharmacologique de l’AMYR et du TRPV4 démontre que ces deux récepteurs sont nécessaires pour que l’amyline humaine oligomère induise ses effets toxiques sur le Ca2+26. En tant que tel, il est probable que l’Aβ médiatise ses effets toxiques sur l’AMYR de manière similaire, bien que ces données n’existent pas encore. L’influx incontrôlé de Ca2+ est lié à un certain nombre de phénomènes pathologiques, notamment la libération vésiculaire incontrôlée, le dysfonctionnement des OS et des mitochondries, l’apoptose, etc. À cette fin, il est probable que le dysfonctionnement cellulaire et le développement d’autres pathologies semblables à celles de la maladie d’Alzheimer qui découlent de la signalisation amyloïde toxique soient médiés à la fois par l’AMYR et le TRPV4. À ce titre, il est nécessaire de discerner les cascades de signalisation qui modulent la relation entre l’AMYR et le TRVP4. De plus, des expériences pharmacologiques sont justifiées, utilisant l’Aβ et le pramlintide sur un large éventail de doses, afin de déterminer les effets de l’Aβ et du pramlintide sur les courants Ca2+, la LTP, la production d’AMPc, et d’autres cascades de signalisation afin de déterminer leurs capacités de signalisation. Ces expériences permettront de combler certains des vides de la littérature actuelle en ce qui concerne l’Aβ et son implication dans les états pathologiques (figure 1).
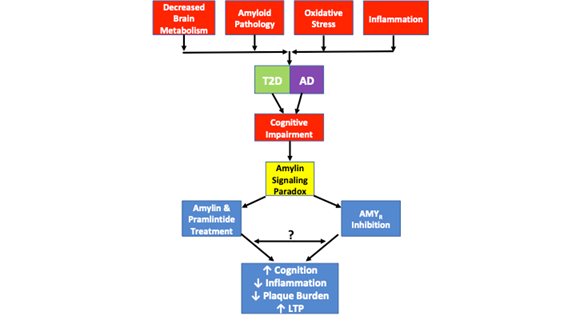
La figure 1. dépeint le paradoxe de signalisation de l’amyline et les similitudes pathologiques observées dans le DT2 et la MA. La diminution du métabolisme cérébral, la pathologie amyloïde, le stress oxydatif et l’inflammation sont autant de caractéristiques pathologiques communes observées dans les deux maladies. Si tous les cas de DT2 ou de MA ne présentent pas chacune de ces caractéristiques pathologiques, tous les cas présentent des troubles cognitifs. Le paradoxe de la signalisation par l’amyline entre en jeu, car des études ont montré que l’inhibition de l’AMYR et l’agonisme de l’AMYR par l’amyline et le pramlintide entraînent une amélioration de la cognition, une diminution de l’inflammation, une réduction de la charge de la plaque et une augmentation de la LTP. Les mécanismes de signalisation régis par l’agonisme et l’antagonisme de l’AMYR n’ont pas encore été entièrement élucidés. Alors que la signalisation de l’amyline est traditionnellement associée à la signalisation de l’AMPc et de la PKA, il n’est pas clair si d’autres cascades sont également activées par l’amyline/pramlintide. De plus, il n’est pas clair si les antagonistes de l’AMYR, les oligomères d’amyline ou l’Aβ signalent par l’intermédiaire de l’AMYR ou s’il existe des similitudes ou une diaphonie entre tous ces ligands de l’AMYR. En tant que tel, un certain nombre d’expériences proposées dans cette revue aideront à élucider davantage la véritable nature de l’AMYR.
Conclusions
La disparité actuelle concernant le rôle de la signalisation de l’amyline dans le cerveau démontre un besoin essentiel d’élucider davantage l’implication de l’amyline à la fois dans la MA et le T2D. Dans le DT2, il est probable qu’aux premiers stades de la maladie, l’amyline envahit le cerveau, forme des oligomères, induit une signalisation aberrante par son récepteur natif et recrute TRPV4 pour induire un influx pathologique de Ca2+ qui entraîne un dysfonctionnement neuronal généralisé qui se manifeste par un OS, une libération vésiculaire incontrôlée et un dysfonctionnement interneuronal, une inflammation et la mort cellulaire qui en résulte. Ce mécanisme pourrait être responsable de la transition initiale entre le cerveau sain et le vieillissement cérébral dans les maladies métaboliques. Ainsi, l’inhibition de l’AMYR ou du TRVP4 à certains moments de la maladie métabolique et aux premiers stades du diabète peut être justifiée pour bloquer les effets toxiques de l’amyline ou de l’Aβ oligomérique. Cependant, des preuves solides suggèrent également que le remplacement de l’amyline par de l’amyline humaine ou du pramlintide réduit la plupart des principales pathologies liées à la MA tout en améliorant la cognition dans des modèles rongeurs de la MA. En tant que tel, le remplacement de la signalisation de l’amyline par l’amyline ou le pramlintide aux stades moyen et avancé du diabète, lorsque la signalisation de l’amyline est perdue en raison de l’agrégation, de l’oligomérisation ou de la perte des cellules β, peut être justifié. À cette fin, il est également nécessaire de discerner la présentation temporelle des événements pathologiques dans le vieillissement cérébral lié au métabolisme et les options thérapeutiques pour les stades précoce, intermédiaire et avancé de la maladie. L’analyse critique et le test de la nature directe et des capacités de signalisation de ces amyloïdes ainsi que la nature thérapeutique des traitements temporels spécifiques peuvent aider à combler le fossé entre les thérapies d’inhibition de l’AMYR et les thérapies de remplacement de l’amyline.
Financement
Le financement de cet article a été assuré par la subvention 1R15AG050292-01A1 des National Institutes of Aging.
- LaFerla FM, Oddo S. Alzheimer’s disease : Aβ, tau et dysfonctionnement synaptique. Trends Mol Med. 2005 ; 11 : 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurologie. 2013 ; 80 : 1778-1783.
- Association As. 2016 Faits et chiffres sur la maladie d’Alzheimer. Alzheimer & Dementia. 2016 ; 12 : 459-509.
- Cummings JL, Morstorf T, Zhong K. Pipeline de développement de médicaments pour la maladie d’Alzheimer : peu de candidats, échecs fréquents. Alzheimers Res Ther. 2014 ; 6 : 37.
- Hyde C, Peters J, Bond M, et al. Évolution des preuves de l’efficacité et du rapport coût-efficacité des inhibiteurs de l’acétylcholinestérase et de la mémantine pour la maladie d’Alzheimer : examen systématique et modèle économique. Vieillissement. 2012 ; 42 : 14-20.
- Howard R, McShane R, Lindesay J, et al. Donépézil et mémantine pour la maladie d’Alzheimer modérée à sévère. N Engl J Med. 2012 ; 366 : 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Taux de glucose et risque de démence. N Engl J Med. 2013 ; 369 : 540-548.
- Loef M, Walach H. Midlife obesity and dementia : meta?analyse and adjusted forecast of dementia prevalence in the United States and China. Obésité. 2013 ; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabète sucré et risque de démence : une méta-analyse d’études observationnelles prospectives. Journal of diabetes investigation. 2013 ; 4 : 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia : a 27 year longitudinal population based study. BMJ. 2005 ; 330 : 1360.
- Vagelatos NT, Eslick GD. Le diabète de type 2 comme facteur de risque de la maladie d’Alzheimer : les facteurs de confusion, les interactions et la neuropathologie associés à cette relation. Epidemiol Rev. 2013 ; 35 : 152-160.
- Debette S, Seshadri S, Beiser A, et al. L’exposition aux facteurs de risque vasculaire à mi-vie accélère le vieillissement cérébral structurel et le déclin cognitif. Neurologie. 2011 ; 77 : 461-468.
- Ginter E, Simko V. Prévalence mondiale et avenir du diabète sucré In Diabetes Springer. 2013 ; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. L’obésité et ses comorbidités. Clin Cornerstone. 1999 ; 2 : 17-31.
- Fédération ID. Atlas du diabète de la FID. Bruxelles : Fédération internationale du diabète. 2013.
- K Dash S. Troubles cognitifs et diabète. Pat récent Endocr Metab Immune Drug Discov. 2013 ; 7 : 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus : a population-based cohort study. Am J Epidemiol. 1997 ; 145 : 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Démence et déclin cognitif dans le diabète de type 2 et les stades prédiabétiques : vers des interventions ciblées. Le lancette Diabète & endocrinologie. 2014 ; 2 : 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabète sucré et maladie d’Alzheimer : pathologie et traitement partagés. Br J Clin Pharmacol. 2011 ; 71 : 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prévalence du diabète et du prédiabète et leurs facteurs de risque chez les adultes bangladais : une enquête nationale. Bull World Health Organ. 2014 ; 92 : 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabète sucré et risque de démence The Rotterdam Study. Neurologie. 1999 ; 53 : 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. L’enzyme de dégradation de l’insuline régule les niveaux d’insuline, de β-protéine amyloïde et du domaine intracellulaire de la protéine précurseur β-amyloïde in vivo. Actes de l’Académie nationale des sciences. 2003 ; 100 : 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. La liaison de l’insuline aux capillaires cérébraux est réduite chez les rats Zucker génétiquement obèses et hyperinsulinémiques. Peptides. 1990 ; 11 : 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987 ; 64 : 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Les niveaux d’insuline sont diminués dans le liquide céphalorachidien des femmes atteintes de la maladie d’Alzheimer prodomale. J Alzheimers Dis. 2010 ; 22 : 405-413.
- Zhang N, Yang S, Wang C, et al. Cible multiple de la hAmyline sur les neurones primaires de l’hippocampe du rat. Neuropharmacologie. 2017 ; 113 : 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mécanismes reliant l’obésité à la résistance à l’insuline et au diabète de type 2. Nature. 2006 ; 444 : 840-846.
- Johnson K, O’Brien T, Jordan K, et al. L’altération de la tolérance au glucose est associée à une augmentation de l’immunoréactivité du polypeptide amyloïde des îlots (IAPP) dans les cellules bêta du pancréas. Le journal américain de pathologie. 1989 ; 135 : 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989 ; 321 : 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. L’activation de l’inflammasome NLRP3 par le polypeptide amyloïde d’îlot fournit un mécanisme pour l’augmentation de l’IL-1β dans le diabète de type 2. Nat Immunol. 2010 ; 11 : 897.
- Wang X, Bao W, Liu J, et al. Marqueurs inflammatoires et risque de diabète de type 2 : une revue systématique et une méta-analyse. Diabetes Care2013 ; 36 : 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. La β-amyloïde d’Alzheimer, l’amyline d’îlot humain et le fragment de protéine prion évoquent des élévations de calcium libre intracellulaire par un mécanisme commun dans une lignée cellulaire neuronale hypothalamique GnRH. J Biol Chem. 2000 ; 275 : 14077-14083.
- Verma N, Ly H, Liu M, et al. Dépôt intraneuronal d’amyline, lésion peroxydative de la membrane et augmentation de la synthèse d’IL-1β dans le cerveau de patients atteints de la maladie d’Alzheimer avec un diabète de type 2 et chez des rats diabétiques HIP. J Alzheimers Dis. 2016 ; 53 : 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Dépôt d’amyline dans le cerveau : un deuxième amyloïde dans la maladie d’Alzheimer. Ann Neurol. 2013 ; 74 : 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. Ensemencement in vivo et ensemencement croisé d’amyloïdose localisée : un lien moléculaire entre le diabète de type 2 et la maladie d’Alzheimer. The American journal of pathology. 2015 ; 185 : 834-846.
- Fu W, Patel A, Jhamandas JH. Le récepteur de l’amyline : une cible physiopathologique commune dans la maladie d’Alzheimer et le diabète sucré. Front Aging Neurosci. 2013 ; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Le peptide β (Aβ) amyloïde active directement le sous-type de récepteur amylin-3 en déclenchant de multiples voies de signalisation intracellulaires. J Biol Chem. 2012 ; 287 : 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Le pramlintide antagonise la dépression de la potentialisation à long terme hippocampique induite par la bêta-amyloïde (Aβ) et l’amyline humaine. Mol Neurobiol. 2017 ; 54 : 748-754.
- Kimura R, MacTavish D, Yang J, et al. La dépression de la potentialisation à long terme hippocampique induite par la bêta-amyloïde est médiée par le récepteur de l’amyline. J Neurosci. 2012 ; 32 : 17401-17406.
- Zhu H, Wang X, Wallack M, et al. L’injection intrapéritonéale du peptide pancréatique amyline réduit puissamment la déficience comportementale et la pathologie amyloïde cérébrale dans des modèles murins de la maladie d’Alzheimer. Mol Psychiatry. 2015 ; 20 : 252.
- Zhu H, Xue X, Wang E, et al. Les ligands du récepteur de l’amyline réduisent la cascade pathologique de la maladie d’Alzheimer. Neuropharmacologie. 2017 ; 119 : 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Effets neuroprotecteurs de l’analogue de l’amyline pramlintide sur la pathogenèse et la cognition de la maladie d’Alzheimer. Neurobiol Aging. 2014 ; 35 : 793-801.
- Zhang Z, Hartmann H, Do VM, et al. La déstabilisation de la β-caténine par des mutations de la préséniline-1 potentialise l’apoptose neuronale. Nature. 1998 ; 395 : 698-702.
- Wang E, Zhu H, Wang X, et al. Le traitement à l’amyline réduit la neuroinflammation et améliore les modèles anormaux d’expression génétique dans le cortex cérébral d’un modèle de souris de la maladie d’Alzheimer. J Alzheimers Dis. 2017 ; 56 : 47-61.
- Singh-Franco D, Robles G, Gazze D. L’acétate de pramlintide injectable pour le traitement du diabète sucré de type 1 et de type 2. Clin Ther. 2007 ; 29 : 535-562.
- Singh?Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes : a systematic review and meta?analysis. Diabète, obésité et métabolisme. 2011 ; 13 : 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Le pramlintide comme complément à l’insulinothérapie améliore le contrôle glycémique et pondéral à long terme chez les patients atteints de diabète de type 2. Diabetes Care. 2003 ; 26 : 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Effets neuroprotecteurs des analogues de l’amyline sur la pathogenèse et la cognition de la maladie d’Alzheimer. J Alzheimers Dis. 2018 ; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Les actions de la protéine β-amyloïde sur les neurones humains sont exprimées par le récepteur de l’amyline. The American journal of pathology. 2011 ; 178 : 140-149.
- Soudy R, Patel A, Fu W, et al. L’AC253 cyclique, un nouvel antagoniste du récepteur de l’amyline, améliore les déficits cognitifs dans un modèle murin de la maladie d’Alzheimer. Alzheimer & Dementia : Recherche translationnelle & Interventions cliniques. 2017 ; 3 : 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Réduction du comportement nociceptif chez les souris knockout du polypeptide amyloïde des îlots (amyline). Mol Brain Res. 1998 ; 63 : 180-183.
- Gingell JJ, Burns ER, Hay DL. Activité du pramlintide, de l’amyline de rat et de l’amyline humaine mais pas de l’Aβ1-42 sur les récepteurs de l’amyline humaine. Endocrinologie. 2014 ; 155 : 21-26.