L’incidence et la prévalence de la maladie des calculs rénaux continuent d’augmenter dans la population générale. Avec une prévalence à vie allant jusqu’à 10%, la néphrolithiase (NL) et la néphrocalcinose (NC) sont donc des fardeaux sanitaires majeurs, notamment dans le monde occidental (1). La NL et la NC sont associées à une morbidité importante et à la progression vers une maladie rénale chronique en raison des récidives, des interventions chirurgicales/endoscopiques répétitives et de l’inflammation concomitante. De manière simplifiée, la formation de calculs rénaux résulte d’un déséquilibre entre les inhibiteurs urinaires (par exemple, le citrate, le magnésium, l’uromoduline et le pyrophosphate) et les promoteurs (par exemple, l’oxalate, le calcium, le phosphate, l’urate et la cystine) de la cristallisation, dépassant la sursaturation avec l’agrégation consécutive, la nucléation et la croissance du calcul à la plaque de Randall (Figure 1). Ce déséquilibre peut être dû à une altération de la manipulation entérale et/ou rénale des promoteurs ou des inhibiteurs, comme une mal-sécrétion entérale d’oxalate ou une mal-réabsorption rénale du calcium (Figure 1).

Figure 1. Déséquilibre des inhibiteurs et promoteurs urinaires de la cristallisation conduisant à la formation de calculs rénaux. La concentration des inhibiteurs et des promoteurs urinaires est influencée et contrôlée par des transporteurs intestinaux et rénaux (axe GI-rein). Ces transporteurs et échangeurs, comme le SLC26A1, sont responsables de la sécrétion et de l’absorption. En ce qui concerne l’oxalate, l’hyperabsorption entérale mais la malécrétion, et/ou l’hypersécrétion rénale mais la malabsorption conduit à des niveaux d’oxalate urinaire dépassant la sursaturation et favorisant ainsi la cristallisation et la formation consécutive de calculs .
L’étiologie sous-jacente de la NL est considérée comme multifactorielle avec une composante environnementale, notamment alimentaire, hormonale et génétique. Dans les études de jumeaux, l’héritabilité des calculs rénaux a été estimée à 56% (3), et jusqu’à deux tiers des personnes hypercalciuriques formant des calculs ont des parents atteints de NL (4). Bien que les calculs rénaux contenant du calcium représentent plus de 80 % de tous les cas, la base génétique de ces calculs reste largement inconnue (5). À l’exception des variants de CLDN14, TRPV5, SLC34A1, ALPL, CASR et UMOD, les études d’association à l’échelle du génome n’ont pas encore révélé de facteurs génétiques substantiels (6-8). Cependant, des allèles de risque ont été identifiés dans des gènes qui se sont également avérés transmettre la maladie sur une base mendélienne, tels que CASR, SLC34A1 et SLC2A9 (9, 10). À ce jour, plus de 30 gènes uniques avec un phénotype défini par l’hérédité mendélienne en ligne chez l’homme ont été identifiés pour être impliqués dans la NL/NC, s’ils sont mutés (tableau 1).

Tableau 1. Gènes, connus pour causer des formes monogéniques de NL/NC.
Les modes d’hérédité dans les formes monogéniques incluent la transmission autosomique-dominante, autosomique-récessive, et liée au chromosome X. De manière intéressante, dans plusieurs de ces gènes, des modes d’hérédité à la fois récessifs et dominants ont été rapportés : SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 et SLC4A1. Alors que la plupart des maladies syndromiques et congénitales graves présentent un mode d’hérédité récessif (Bartter, Lowe, Dent, FHHNC et acidose tubulaire rénale distale avec surdité de perception), les affections plus légères sont plutôt associées à des mutations dans des gènes dominants. La majorité des protéines codées constituent des transporteurs de solutés rénaux (par exemple, SLC34A1, SLC34A3 et SLC9A3R1), mais des canaux de chlorure (CLCN5), des protéines de jonction serrée (par exemple, CLDN16/CLDN19) et des enzymes métaboliques (par exemple, AGXT, APRT et CYP24A1) se sont révélés défectueux chez les patients atteints de NL/NC. Par conséquent, le défaut sous-jacent est principalement situé dans le système tubulaire du rein lui-même et peut donc être attribué à une tubulopathie. A l’inverse, des conditions a priori extrarénales, comme dans l’hyperoxalurie primaire (PH) où le dysfonctionnement d’enzymes hépatiques (AGXT, GRHPR, et HOGA1) provoque une accumulation d’oxalate avec une affection rénale secondaire, sont des causes concevables de NL/NC. Bien que l’on pense que chaque phénotype de maladie représente une entité relativement rare, les causes monogéniques peuvent expliquer un nombre significatif de patients par leur large hétérogénéité génétique (42). Outre l’hétérogénéité génétique, il existe également une variation allélique, où les variants tronquants entraînent plutôt une perte de fonction et les variants faux sens (hypomorphes) peuvent provoquer des défauts plutôt subtils, qui peuvent être surveillés cliniquement, en particulier chez les adultes qui forment des calculs. Un autre phénomène récemment apprécié concerne les effets du dosage des gènes dans plusieurs des gènes de calculs rénaux susmentionnés. Dans le cas de SLC34A3, par exemple, qui code pour l’un des principaux transporteurs de phosphate dans le tubule proximal (NaPiIIc), il a été démontré que les individus hétérozygotes ne peuvent plus être considérés comme de simples porteurs sains, car ils présentent des calcifications rénales et/ou des manifestations osseuses beaucoup plus fréquemment que les individus de type sauvage, mais toujours à un degré moindre que les individus bialéliques (homozygotes et hétérozygotes composés) (43). Des observations similaires ont été rapportées pour les familles présentant des mutations du CYP24A1 (44). La contribution des troubles monogéniques à la prévalence globale de la maladie des calculs rénaux n’a pas été étudiée de manière exhaustive dans le passé. En particulier, on manque de preuves génétiques basées sur un large dépistage d’une multitude de gènes causaux dans de grandes cohortes de patients. Les tests génétiques complets étaient jusqu’à présent trop coûteux et inefficaces. Pour la plupart des personnes atteintes de NL/NC, l’analyse de mutation pour un défaut génétique causal n’a donc pas été accessible, malgré le fait que la connaissance de la cause moléculaire de la NL/NC peut avoir des conséquences importantes pour le pronostic, la prophylaxie et/ou le traitement. Seules des estimations approximatives ont été dérivées d’études d’observation clinique : sur la base d’une énorme collection de données d’analyse de la composition des calculs, il a été conclu que les causes monogéniques ne dépassent pas 9,6% chez les enfants et 1,6% chez les adultes (45). Au cours de la dernière décennie, cependant, cette situation a commencé à changer, avec l’avènement des techniques de séquençage à haut débit.
Analyse de mutation à haut débit chez les patients atteints de NL/NC
Pour étudier chez les patients atteints de calculs rénaux la présence de mutations pathogènes dans des gènes de maladie connus, nous avons établi un panel de gènes basé sur la PCR multiplex microfluidique et le séquençage NextGen consécutif (Fluidigm™/NGS) (46, 47).
Dans une « étude pilote », nous avons recruté consécutivement 268 individus génétiquement non résolus provenant de cliniques typiques de calculs rénaux ; 102 probands pédiatriques et 166 adultes. Nous avons ainsi identifié 50 variants délétères dans 14 des 30 gènes analysés, ce qui a permis de poser un diagnostic moléculaire dans 15 % des cas. Dans le sous-groupe pédiatrique, nous avons détecté une mutation causale dans 21 % des cas, tandis que chez les adultes, des variants délétères étaient présents dans 11 % des cas (figure 2A) (48). Les mutations dans le gène de la cystinurie SLC7A9 ont été trouvées le plus fréquemment dans la cohorte adulte (Figure 2B). Deux études de suivi ont permis de confirmer ces résultats. Premièrement, dans une cohorte exclusivement pédiatrique de 143 patients atteints de NL/NC, 17 % des cas étaient expliqués par des mutations dans 14 gènes différents (49). Deuxièmement, dans une cohorte de 51 familles dont l’âge de la manifestation de la NL/NC était inférieur à 25 ans, une WES ciblée a permis de détecter une cause génétique dans près de 30 % des cas (50). Comme on pouvait s’y attendre, les mutations récessives étaient plus fréquemment trouvées chez les nouveau-nés et dans les cas de maladie congénitale, alors que les conditions dominantes se manifestaient généralement plus tard dans la vie. Ces données indiquent que la maladie génétique des calculs rénaux est une condition sous-diagnostiquée, malgré le fait que le diagnostic moléculaire influencera potentiellement le pronostic, la prophylaxie et/ou le traitement. Une limitation qui mérite d’être mentionnée, cependant, est un biais de sélection potentiel dû au recrutement dans des cliniques spécialisées dans les calculs rénaux dans les trois études susmentionnées.

Figure 2. Analyse des mutations de 30 gènes monogéniques connus de la néphrolithiase (NL)/néphrocalcinose (NC) chez 268 patients atteints de NL/NC. (A) Fraction des causes monogéniques dans la sous-cohorte pédiatrique et adulte. (B) Nombre de causes monogéniques pour l’ensemble des gènes (en rouge, les adultes ; en orange, les patients pédiatriques). Il convient de noter que SLC7A9 a été trouvé le plus fréquemment muté, en particulier chez les jeunes adultes.
Identification de nouveaux gènes de maladies humaines par une approche de gènes candidats
L’analyse de mutation à haut débit est également utilisée pour cribler les variantes pathogènes dans divers gènes candidats. L’une des découvertes récentes les plus intéressantes a été la découverte de mutations humaines dans SLC26A1 (32). Depuis la première description de la formation de calculs rénaux à base de Ca-oxalate (CaOx) et de NC chez des souris Slc26a1 (Sat1)-knockout par Dawson et al. en 2010, SLC26A1 est un gène candidat NL de bonne foi (51). SLC26A1 code pour un échangeur d’anions exprimé au niveau de la membrane basolatérale des tubules rénaux proximaux, de l’iléon et du jéjunum. Par conséquent, en utilisant une approche par gène candidat, des variantes pathogènes ont été identifiées chez des humains ayant des antécédents de CaOx-NL précoce, à savoir deux individus non apparentés présentant des variantes bialéliques faux-sens (32). Sur le plan fonctionnel, la pathogénicité des variants identifiés a été démontrée in vitro, entraînant un mauvais trafic intracellulaire et une activité de transport altérée (32). La défectuosité de SLC26A1 constitue donc une nouvelle cause de CaOx-NL et devrait être prise en compte lors du dépistage des causes de formation récurrente de calculs de CaOx chez les individus.
Nouveau registre clinique de patients pour la maladie héréditaire des calculs rénaux
La plupart des données épidémiologiques sur la prévalence croissante dans les pays occidentaux proviennent de bases de données américaines. Bien qu’il y ait un besoin urgent, les bases de données européennes centralisées ne sont pas disponibles à l’heure actuelle. Comme les études génétiques susmentionnées sur la prévalence de la maladie héréditaire des calculs rénaux ont été réalisées avec de petites cohortes provenant de centres spécialisés en Europe et aux États-Unis, une transposition à la situation générale en Europe n’est pas valable. Alors qu’aux États-Unis, le Rare Kidney Stone Consortium constitue une plateforme qui intègre et coordonne les activités d’enregistrement, de science fondamentale et de recherche clinique pour des maladies rares telles que la cystinurie, la PH, la déficience en APRT, la maladie de Dent et de Lowe, aucune collecte de données comparable sur les patients atteints de la maladie héréditaire des calculs rénaux n’a été mise en place ni en Europe ni en Allemagne à ce jour. En collaboration avec le registre européen existant de la PH, OxalEurope (Prof. Bernd Hoppe, Université de Bonn), et grâce à un financement de la Deutsche Forschungsgemeinschaft et de la Else Kröner-Fresenius Stiftung, nous avons récemment établi un « Registre pour la maladie héréditaire des calculs rénaux » à l’Université de Leipzig. Ce registre est soutenu au niveau national par les sociétés allemandes de néphrologie adulte (DGfN) et de néphrologie pédiatrique (GPN). Il est également inscrit au registre allemand des essais cliniques (DRKS-ID : DRKS00012891). En tant qu’élément fondamental du recrutement de l’étude, l’analyse de mutation à haut débit pour les gènes de calculs rénaux connus et nouveaux est proposée à titre de recherche pour les patients sans diagnostic moléculaire établi mais dont le tableau clinique indique une susceptibilité génétique sous-jacente : par exemple, âge d’apparition précoce (<40 ans), antécédents familiaux positifs, phénotypes indicatifs tels que NC, cystinurie ou RTA, et NL sévèrement récurrente (>3×) (tableau 2). Alors que les patients avec un diagnostic génétique déjà établi sont généralement recrutés, les cas avec des causes secondaires de NL/NC, comme les tumeurs malignes, la sarcoïdose et l’hyperparathyroïdie primaire, ne sont pas inclus dans l’analyse génétique. Pour recruter activement des patients, un centre clinique doit généralement obtenir l’approbation du conseil d’examen institutionnel local ; un processus pour lequel nous offrons notre aide et notre assistance en fournissant des modèles respectifs. Une fois l’approbation éthique obtenue, le formulaire de consentement et les fiches de données cliniques (par exemple, le questionnaire clinique) peuvent être téléchargés sur le site Web de notre registre (http://www.mks-registry.net). Afin de garantir un phénotypage clinique complet, nous demanderons des informations importantes sur les patients, telles que l’origine ethnique, la consanguinité, les antécédents familiaux, l’âge d’apparition, la récurrence (définie comme tout nouvel épisode de calculs rénaux), la consommation quotidienne de liquides, les interventions chirurgicales et l’implication extrarénale, entre autres. Les documents peuvent être remplis par le patient avec l’aide du médecin qui l’a inscrit. En outre, les paramètres biochimiques sériques, y compris la créatinine, le DFGe, la PTH, la vitamine D, les électrolytes, l’acide urique et l’analyse d’urine (pH, calcium, phosphate, magnésium, acide urique, citrate, oxalate et cystine, de préférence à partir d’une urine de 24 heures, sinon d’une urine ponctuelle), ainsi que les données sur l’analyse de la composition des calculs seront demandés lors de l’inscription. Compte tenu du fait que la collecte d’urine de 24 heures et l’analyse de la composition des calculs ne sont pas systématiquement effectuées dans toutes les institutions, nous incluons ces paramètres selon leur disponibilité. Des visites de suivi clinique des patients inscrits tous les deux ans sont souhaitables mais pas obligatoires. Après l’inscription, les centres cliniques recruteurs recevront un login personnalisé pour saisir les données des patients via le site web de notre registre (http://www.mks-registry.net). Alternativement, nous proposons de saisir les données par voie électronique lorsqu’elles nous sont envoyées sur papier. Les données saisies seront stockées sur un serveur sécurisé et les centres cliniques participants pourront y accéder pour consulter les données de leurs propres patients. Les sites web suivants fournissent des informations supplémentaires :
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
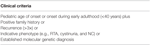
Tableau 2. Critères d’inclusion pour l’analyse des mutations dans le registre clinique des patients.
En résumé, la maladie des calculs rénaux est une affection de plus en plus prévalente, cliniquement hétérogène et mal comprise, notamment ses moteurs génétiques. Comme l’indique une série d’études récentes, la prévalence des affections monogéniques est très probablement sous-estimée. La mise en place d’un registre centralisé des patients souffrant de calculs rénaux héréditaires contribuera à combler, du moins en partie, le vaste fossé des connaissances sur la génétique des calculs rénaux. Dans ce contexte, les registres cliniques sont des sources précieuses pour plusieurs raisons : premièrement, la délimitation de meilleures associations phénotype-génotype sera cruciale pour une stratification plus précise des patients dans les futures études de recherche clinique. Deuxièmement, l’identification de nouveaux gènes de maladie avec de nouveaux mécanismes de maladie diminuera le fossé de l’étiologie inconnue de la NL/NC ; et troisièmement, le déchiffrage de nouvelles cibles moléculaires aide à ouvrir la voie au développement de médicaments de prévention des récidives dans les familles gravement touchées.
Déclaration d’éthique
Cette étude a été réalisée conformément aux recommandations de la « Ethikkommission an der Medizinischen Fakultät der Universität Leipzig » avec le consentement éclairé écrit de tous les sujets. Tous les sujets ont donné leur consentement éclairé par écrit, conformément à la Déclaration d’Helsinki. Le protocole a été approuvé par la « Ethikkommission an der Medizinischen Fakultät der Universität Leipzig. »
Contributions des auteurs
JH a conçu et rédigé le manuscrit. BH, AS, LM et RS ont édité le manuscrit et construit l’infrastructure du registre qui est présentée au lecteur.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Financement
La recherche est financée par des subventions de projet de la DFG (HA 6908/2-1) et de l’EKFS (2016_A52) à JH. Ce travail a également été soutenu par le ministère fédéral de l’éducation et de la recherche (BMBF), Allemagne, FKZ : 01EO1501 à JH.
1. Scales CD Jr, Smith AC, Hanley JM, Saigal CS. Prévalence des calculs rénaux aux États-Unis. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Mise à jour sur la néphrolithiase : programme de base 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. Une étude de jumeaux sur les influences génétiques et alimentaires sur la néphrolithiase : un rapport du Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Génétique de la néphrolithiase hypercalciurique : maladie des calculs rénaux. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Progrès dans la compréhension de la génétique de la néphrolithiase calcique. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576.
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Les variantes de séquence dans le gène CLDN14 sont associées aux calculs rénaux et à la densité minérale osseuse. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Variants communs et rares associés aux calculs rénaux et aux traits biochimiques. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 influence les concentrations d’acide urique avec des effets prononcés spécifiques au sexe. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Les mutations du gène SLC2A9 du transporteur de glucose 9 provoquent une hypouricémie rénale. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identification et caractérisation d’un gène avec des substitutions de base associées au phénotype de l’hypercalciurie absorbante et à une faible densité osseuse spinale. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Adénine phosphoribosyltransférase humaine. Identification de mutations alléliques au niveau des nucléotides comme cause de la déficience complète de l’enzyme. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Des mutations dans ATP6N1B, codant pour une nouvelle sous-unité 116-kD de la pompe à protons vacuolaire du rein, provoquent une acidose tubulaire rénale distale récessive avec une audition préservée. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutations dans le gène codant pour la sous-unité B1 de la H+-ATPase causant une acidose tubulaire rénale avec surdité de perception. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Le syndrome de déficience en anhydrase carbonique II dans une famille belge est causé par une mutation ponctuelle au niveau d’un résidu histidine invariant (107 His→Tyr) : structure complète du gène humain normal de l’AC II. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A family syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. A common molecular basis for three inherited kidney stone diseases. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutations dans le gène du canal chlorure, CLCNKB, cause Bartter’s syndrome type III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, une protéine de jonction serrée rénale requise pour la résorption paracellulaire de Mg2+. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Les mutations du gène de jonction serrée claudine 19 (CLDN19) sont associées à une perte de magnésium rénal, à une insuffisance rénale et à une atteinte oculaire grave. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations dans CYP24A1 et hypercalcémie infantile idiopathique. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nephrocalcinose (syndrome rénal de l’émail) causée par des mutations autosomiques récessives de FAM20A. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Le gène codant pour l’hydroxypyruvate réductase (GRHPR) est muté chez les patients atteints d’hyperoxalurie primaire de type II. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. La mutation HNF4A R76W provoque un syndrome de Fanconi dominant atypique en plus d’un phénotype de cellule β. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identification de 17 mutations indépendantes responsables de la déficience en hypoxanthine-guanine phosphoribosyltransférase (HPRT) humaine. Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Hétérogénéité génétique du syndrome de Bartter révélée par des mutations dans le canal K+, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, syndrome de Bartter anténatal transitoire et mutations MAGED2. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Marqueurs flanquants étroitement liés pour le syndrome oculocérébral de Lowe, avec application à l’évaluation des porteurs. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Les mutations de SLC26A1 causent la néphrolithiase. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Identification moléculaire d’un échangeur d’anions d’urate rénal qui régule les niveaux d’urate sanguins. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinurie causée par des mutations dans rBAT, un gène impliqué dans le transport de la cystine. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identification de deux mutations du gène de la xanthine déshydrogénase humaine responsables de la xanthinurie classique de type I. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Hildebrandt F. Maladies rénales génétiques. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Les mutations de SLC34A3/NPT2c sont associées aux calculs rénaux et à la néphrocalcinose. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Phénotypes cliniques et biochimiques d’adultes présentant des mutations monoalléliques et bialéliques de CYP24A1 : preuve d’un effet de dose du gène. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Récents acquis diagnostiques et thérapeutiques. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification de 99 nouvelles mutations dans une cohorte mondiale de 1 056 patients atteints d’une ciliopathie liée à la néphronophthèse. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Quatorze gènes monogéniques sont responsables de 15 % de la néphrolithiase/néphrocalcinose. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prévalence des causes monogéniques chez les patients pédiatriques atteints de néphrolithiase ou de néphrocalcinose. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Le séquençage de l’exome entier détecte fréquemment une cause monogénique dans la néphrolithiase et la néphrocalcinose à début précoce. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar
.