Alexei Drummond, Andrew Rambaut, Remco Bouckaert, et Walter Xie
1 Introduction
Ce tutoriel présente le logiciel BEAST pour l’analyse bayésienne de l’évolution à travers un tutoriel simple. Le tutoriel implique la co-estimation d’une phylogénie de gènes et des temps de divergence associés en présence d’informations de calibration provenant de preuves fossiles.
Vous aurez à votre disposition les logiciels suivants :
- BEAST – ce paquet contient le programme BEAST, BEAUti, TreeAnnotator et d’autres programmes utilitaires. Ce tutoriel est écrit pour BEAST v2.2.x, qui prend en charge les partitions multiples. Il est disponible en téléchargement à partir de http://www.beast2.org/.
- Tracer – ce programme est utilisé pour explorer la sortie de BEAST (et d’autres programmes MCMC bayésiens). Il résume graphiquement et quantitativement les distributions des paramètres continus et fournit des informations de diagnostic. Au moment de la rédaction de cet article, la version actuelle est v1.6. Elle peut être téléchargée à partir de
http://tree.bio.ed.ac.uk/software/. - FigTree – il s’agit d’une application permettant d’afficher et d’imprimer des phylogénies moléculaires, en particulier celles obtenues à l’aide deBEAST. Au moment de la rédaction de cet article, la version actuelle est la v1.4.2. Elle peut être téléchargée à partir de http://tree.bio.ed.ac.uk/software/.
Ce tutoriel vous guidera dans l’analyse d’un alignement de séquences échantillonnées chez douze espèces de primates (voir Figure 1). L’objectif est d’estimer la phylogénie, le taux d’évolution sur chaque lignée et les âges des divergences non calibréesancestrales.
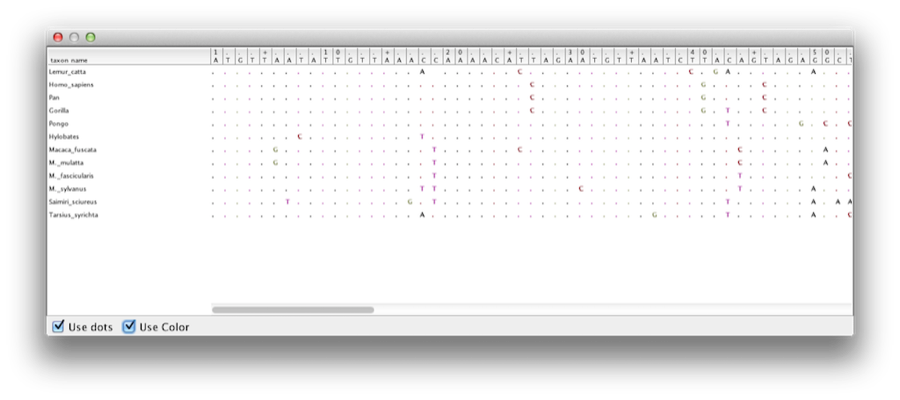
Figure 1 : Partie de l’alignement pour les primates.
La première étape consistera à convertir un fichier NEXUS avec un bloc DATA ou CHARACTERS en un fichier d’entrée XML BEAST. Cela se fait à l’aide du programme BEAUti (qui signifie Bayesian Evolutionary Analysis Utility). Il s’agit d’un programme convivial permettant de définir le modèle évolutif et les options de l’analyse MCMC. La deuxième étape consiste à exécuter BEAST en utilisant le fichier d’entrée généré par BEAUTi, qui contient les données, le modèle et les paramètres d’analyse. La dernière étape consiste à explorer la sortie de BEAST afin de diagnostiquer les problèmes et de résumer les résultats.
2 BEAUti
Le programme BEAUti est un programme convivial permettant de définir les paramètres du modèle pour BEAST. Exécutez BEAUti en double-cliquant sur son icône. Une fois lancé, BEAUti sera similaire quel que soit le système informatique sur lequel il est exécuté. Pour ce tutoriel, la version Mac OS X est utilisée dans les figures, mais les versions Linux et Windows auront la même disposition et les mêmes fonctionnalités.
2.1 Chargement du fichier NEXUS
Pour charger un alignement au format NEXUS, il suffit de sélectionner l’option Importer l’alignement… dans le menu Fichier, ou de faire glisser le fichier au milieu du panneau Partitions.
Le fichier exemple appelé primate-mtDNA.nex est disponible dans le répertoire examples/nexus/ pour Mac et Linux et examples/nexus/ pour Windows à l’intérieur du répertoire où BEAST a été installé.Ce fichier contient un alignement de séquences de 12 espèces de primates.
Une fenêtre d’ajout de partition (Figure 2) apparaîtrait si le paquetage correspondant est installé. Si vous utilisez BEAST 2 « pur », vous pouvez passer au paragraphe suivant. Sinon, sélectionnez Ajouter un alignement et cliquez sur OK pour continuer.
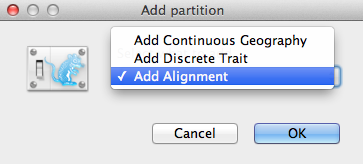
Figure 2 : Fenêtre Ajouter une partition (n’apparaît que si les paquets connexes sont installés).
S’il y a des chevauchements de codage dans les partitions, la fenêtre de message d’avertissement (Figure 3) apparaîtra. Lisez et cliquez sur OK pour continuer.
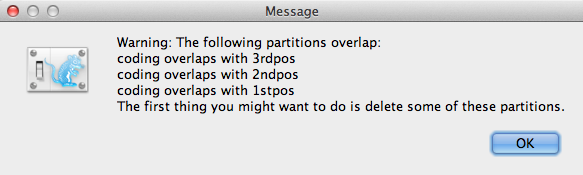
Figure 3 : Fenêtre de message d’avertissement (n’apparaît que s’il y a des chevauchements de codage dans les partitions).
Une fois chargées, des partitions de cinq caractères sont affichées dans le panneau principal (figure 4). L’alignement est divisé en une partie codante de la protéine et une partie non codante,et la partie codante est divisée en positions de codon 1, 2 et 3. Vous devez supprimer la partition ‘coding’ avant de passer à l’étape suivante car elle se réfère aux mêmes nucléotides que les partitions ‘1stpos’, ‘2ndpos’ et ‘3rdpos’. Pour supprimer la partition ‘codage’, sélectionnez la ligne et cliquez sur le bouton ‘-‘ en bas du tableau. Vous pouvez visualiser l’alignement en double cliquant sur la partition.
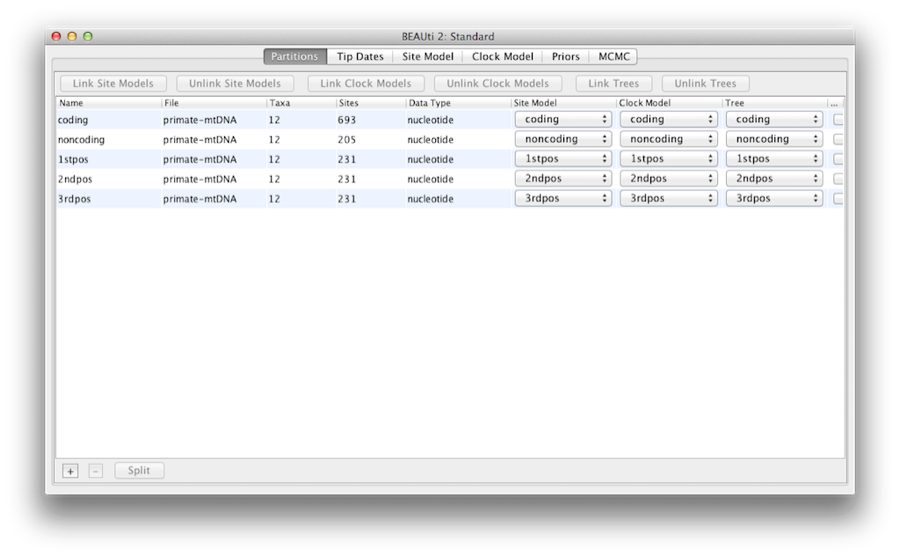
Figure 4 : Une capture d’écran de l’onglet des données dans BEAUti. Cette capture d’écran et toutes les suivantes ont été réalisées sur un ordinateur Apple fonctionnant sous Mac OS X et seront légèrement différentes sur d’autres systèmes d’exploitation.
Lier/dissocier les modèles de partition
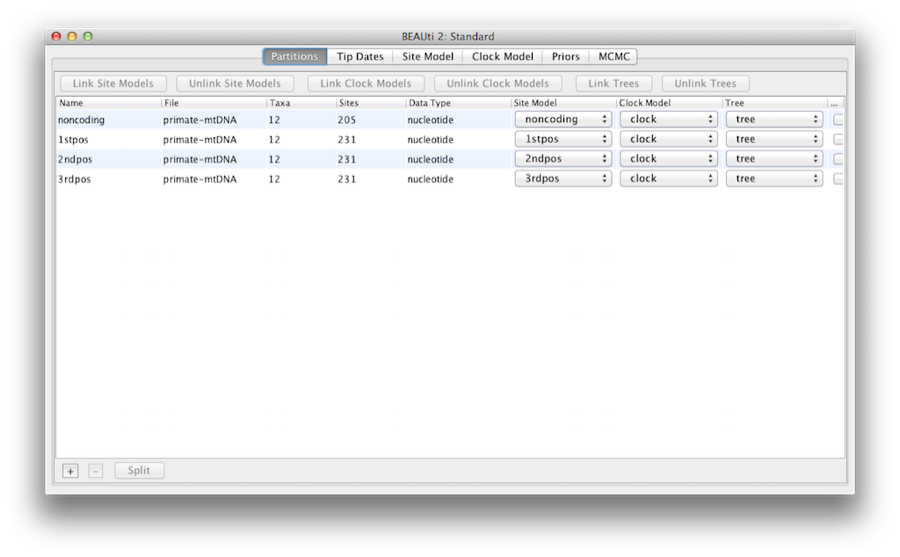
Figure 5 : Une capture d’écran de l’onglet Partitions dans BEAUti après avoir lié et renommé le modèle d’horloge et l’arbre.
Puisque les séquences sont liées (c’est-à-dire qu’elles proviennent toutes du génome mitochondrialqui n’est pas censé subir de recombinaison chez les oiseaux et les mammifères), elles partagent le même ancêtre, donc les partitions devraient partager le même arbre temporel dans le modèle. Pour des raisons de simplicité, nous supposerons également que les partitions partagent le même taux d’évolution pour chaque branche, et donc le même « modèle d’horloge ».Nous limiterons notre modélisation de l’hétérogénéité du taux à l’hétérogénéité entre sites au sein de chaque partition, et nous permettrons également aux partitions d’avoir des taux d’évolution moyens différents.
Donc, à ce stade, nous devrons relier le modèle d’horloge et l’arbre. Dans le panneau Partitions, sélectionnez les quatre partitions du tableau (ou aucune, par défaut toutes les partitions sont concernées) et cliquez sur le bouton Lier les arbres, puis sur le bouton Lier les modèles d’horloge (voir Figure 5). Cliquez ensuite sur le premier menu déroulant de la colonne Clock Model et renommez le modèle d’horloge partagé en « clock ». De même, renommez l’arbre partagé en ‘tree’. Cela rendra les options suivantes et les fichiers journaux générés plus faciles à lire.
2.2 Configuration du modèle de substitution
L’étape suivante consiste à configurer le modèle de substitution. Ensuite, sélectionnez l’onglet Modèles de site en haut de la fenêtre principale (nous sautons l’onglet Tip Dates puisque tous les taxons proviennent d’échantillons contemporains). Ceci révélera les paramètres du modèle d’évolution pour BEAST. Les options disponibles dépendent du fait que les données sont des nucléotides, ou des acides aminés, des données binaires, ou des données générales. Les paramètres qui apparaîtront après avoir chargé l’alignement de nucléotides de primates seront les valeurs par défaut pour les données de nucléotides, nous devons donc effectuer quelques modifications.
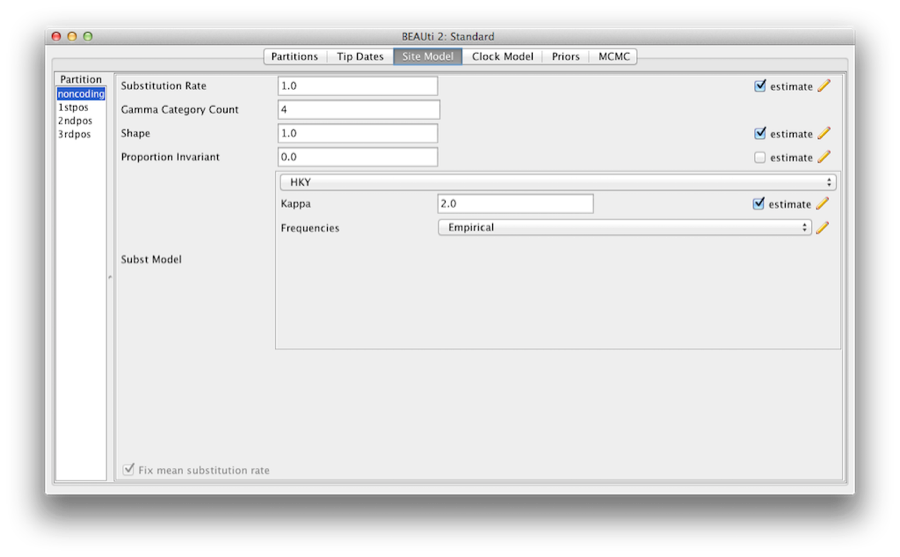
Figure 6 : Une capture d’écran de l’onglet du modèle de site dans BEAUti.
La plupart des modèles devraient vous être familiers. Tout d’abord, réglez le compte de la catégorie Gamma à 4, puis cochez la case » estimation » pour le paramètre Forme. Cela permettra de modéliser la variation du taux entre les sites de chaque partition. Notez que 4 à 6 catégories fonctionnent suffisamment bien pour la plupart des ensembles de données, alors qu’avoir plus de catégories prend plus de temps à calculer pour peu d’avantages supplémentaires. Nous laissons l’entrée Proportion Invariante définie à zéro.
Puis sélectionnez HKY dans le menu déroulant Modèle de substitution. Idéalement, il faudrait sélectionner un modèle de substitution qui s’adapte le mieux aux données pour chaque partition, mais ici, par souci de simplicité, nous utilisons HKY pour toutes les partitions. En outre, sélectionnez Empirique dans le menu déroulant Fréquences. Cela fixera les fréquences aux proportions observées dans les données (pour chaque partition individuellement, une fois que nous aurons dissocié les modèles de site). Cette approche signifie que nous pouvons obtenir un bon ajustement aux données sans estimer explicitement ces paramètres. Nous le faisons ici simplement pour rendre les fichiers journaux un peu plus courts et plus lisibles dans les parties ultérieures de l’exercice.
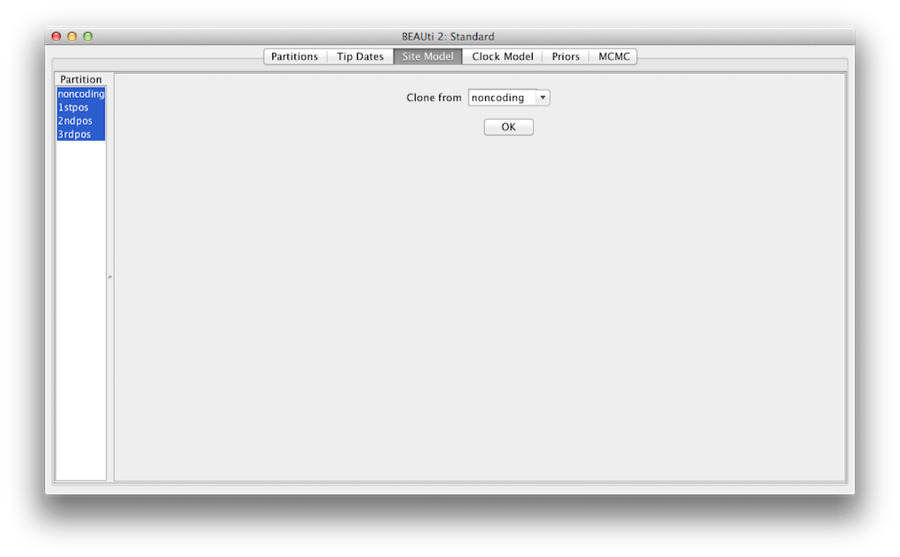
Figure 7 : configuration du clone d’un modèle de site à d’autres.
Enfin, cochez la case ‘estimation’ pour le paramètre Taux de substitution et cochez la case Fixer le taux de mutation moyen. Cela permettra aux partitions individuelles d’avoir leurs taux relatifs estimés pour les modèles de sites non liés (Figure 6).
Enfin, maintenez la touche ‘shift’ pour sélectionner tous les modèles de sites du côté gauche, et cliquez sur OK pour cloner le paramètre de non codage en 1stpos, 2ndpos et 3rdpos (Figure 7). Passez par chaque modèle de site, comme vous pouvez le voir, leurs configurations sont les mêmes maintenant.
2.3 Réglage du modèle d’horloge
L’étape suivante consiste à sélectionner l’onglet Clock Models en haut de la fenêtre principale. C’est ici que nous sélectionnons le modèle d’horloge moléculaire. Pour cet exercice, nous allons laisser la sélection à la valeur par défaut d’une horloge moléculaire stricte, car ces données sont très proches de l’horloge, et n’ont pas besoin de variation de taux entre les branches pour être incluses dans le modèle.
Pour tester la conformité à l’horloge, vous pouvez (i) exécuter l’analyse avec un modèle d’horloge relaxé et vérifier combien de variation entre les taux sont impliqués par les données (voir le coefficient de variation pour plus d’informations à ce sujet), ou(ii) effectuer une comparaison de modèle entre une horloge stricte et relaxée en utilisant pathsampling, ou (iii) utiliser un modèle d’horloge locale aléatoire qui considère explicitement si chaque branche de l’arbre a besoin de son propre taux de branchement.
2.4 Priors
L’onglet Priors permet de spécifier les priors pour chaque paramètre du modèle. Les sélections de modèle effectuées dans les onglets du modèle de site et du modèle d’horloge, entraînent l’inclusion de divers paramètres dans le modèle, et ceux-ci sont indiqués dans l’onglet des prieurs (voir Figure 8).
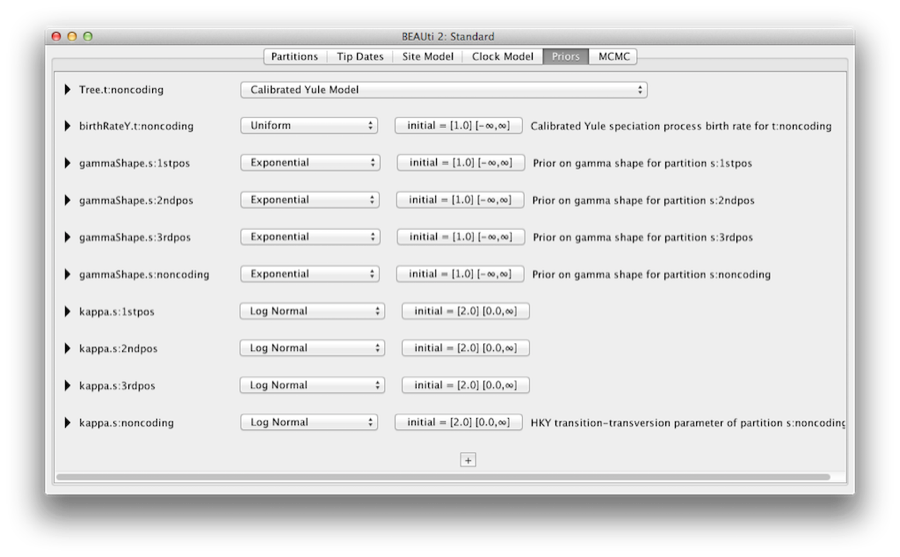
Figure 8 : Une capture d’écran de l’onglet des prieurs dans BEAUti.
Nous spécifions également ici que nous souhaitons utiliser le modèle de Yule calibré comme antériorité de l’arbre. Le modèle de Yule est un modèle simple de spéciation qui est généralement plus approprié lorsque l’on considère des séquences provenant d’espèces différentes. Sélectionnez le modèle de Yule calibré dans le menu déroulant des antériorités de l’arbre.
2.4.1 Définition du nœud de calibrage
Nous devons maintenant spécifier une distribution antérieure sur le nœud calibré, en fonction de notre connaissance des fossiles. Cette opération est connue sous le nom de calibrage de notre arbre. Pour définir une priorité supplémentaire, appuyez sur le petit bouton + sous la liste des priorités. S’il n’est pas visible dans votre vue, veuillez faire défiler le panneau jusqu’en bas pour trouver le bouton +. Vous verrez apparaître une boîte de dialogue qui vous permettra de définir un sous-ensemble de taxons dans l’arbre phylogénétique. Une fois que vous avez créé un ensemble de taxons, vous serez en mesure d’ajouter des informations de calibration pour son plus récent ancêtre commun (MRCA) plus tard.
Nommez l’ensemble de taxons en remplissant l’entrée de l’étiquette de l’ensemble de taxons. Appelez-le humain-chimpanzé, puisqu’il contiendra les taxons pour Homo sapiens et Pan. Dans la liste ci-dessous, vous verrez les taxons disponibles. Sélectionnez tour à tour chacun des deux taxons et appuyez sur le bouton fléché > >. (Comme il s’agit d’un nœud calibré à utiliser en conjonction avec l’antécédent Yule calibré, la monophylie doit être appliquée, donc cochez la case Monophylétique. Cela contraindra la topologie de l’arbre de sorte que le groupement humain-chimpanzé soit maintenu monophylétique au cours de l’analyse MCMC.
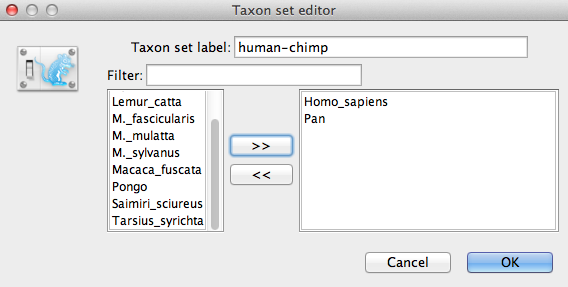
Figure 9 : éditeur d’ensemble de taxons dans BEAUti.
Pour coder les informations de calibration, nous devons spécifier une distribution pour le MRCA de l’homme-chimpanzé.Sélectionnez la distribution Log-normale dans le menu déroulant à droite de l’homme-chimpanzé nouvellement ajouté.prior. Cliquez sur le triangle noir et un graphique de la fonction de densité de probabilité apparaîtra, ainsi que les paramètres de la distribution log-normale. Nous allons définir M=1,78 et S=0,085, ce qui spécifiera une distribution centrée sur environ 6 millions d’années avec un écart-type d’environ 0,5 million d’années. Cela donnera une plage centrale de probabilité de 95 % couvrant 5 à 7 millions d’années. Cela correspond à peu près à l’estimation consensuelle actuelle de la date de l’ancêtre commun le plus récent des humains et des chimpanzés (Figure 10).
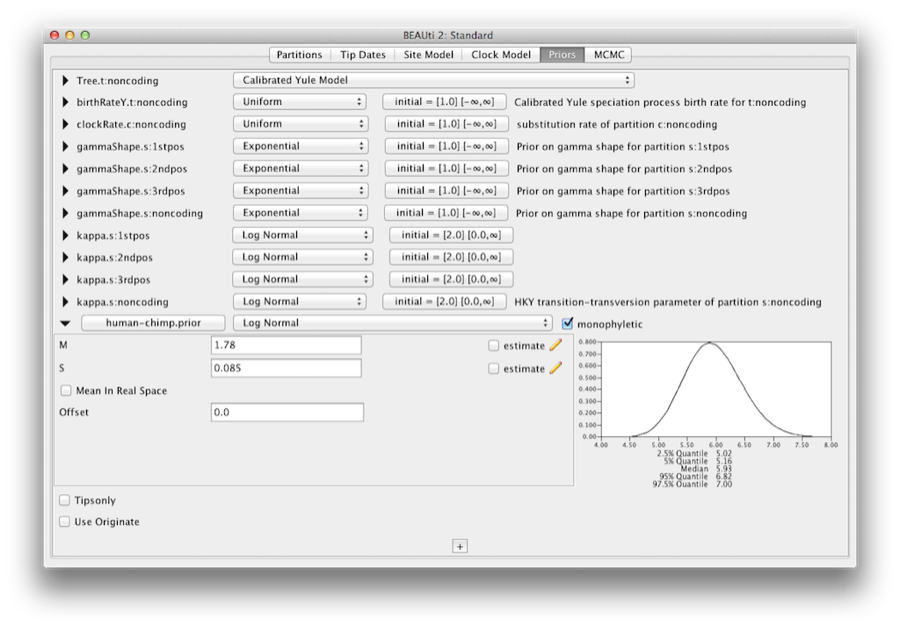
Figure 10 : Une capture d’écran des options de priorisation de calibration dans le panneau Priors de BEAUti.
Nous devons nous convaincre que les prieurs affichés dans le panneau priors reflètent réellement les informations préalables dont nous disposons sur les paramètres du modèle. Enfin, nous allons également spécifier quelques prieurs diffus « non informatifs » mais appropriés sur le taux global de l’horloge moléculaire (clockRate) et le taux de spéciation (birthRateY) de l’arbre de Yule prior. Pour chacun d’entre eux, sélectionnez Gamma dans le menu déroulant et, à l’aide du bouton fléché, développez la vue pour révéler les paramètres de la priorité Gamma. Pour le taux d’horloge et le taux de naissance de Yule, définissez le paramètre Alpha(shape) à 0,001 et le paramètre Beta (scale) à 1000 (Figure 11).
Par défaut, chacun des paramètres de forme gamma a une priordistribution exponentielle avec une moyenne de 1. Cela implique (voir Figure 3.7) que nous nous attendons à une variation somerate. Par défaut, les paramètres kappa du modèle HKY ont une distribution préalable lognormale(1,1.25), ce qui correspond largement aux données empiriques sur la gamme de valeurs réalistes pour les biais de transition/transversion. Ces antériorités par défaut sont conservées car elles conviennent à cette analyse particulière.
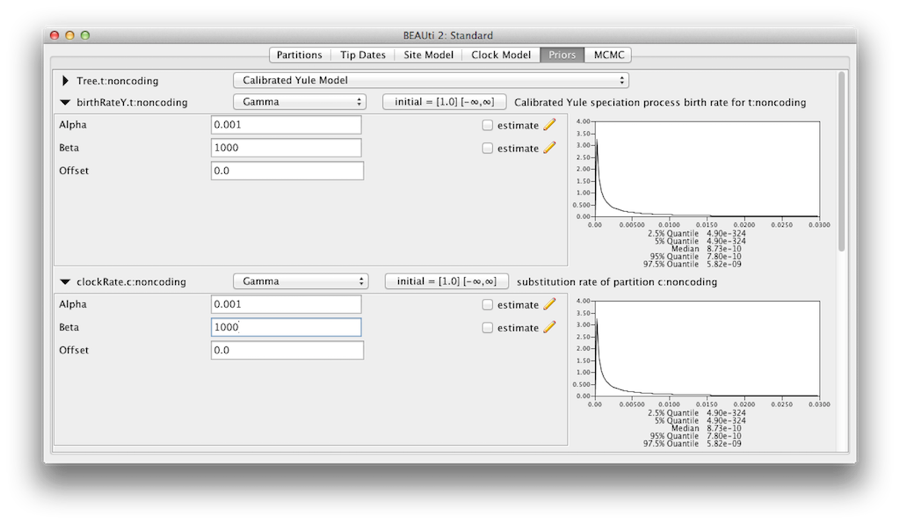
Figure 11 : Antériorité gamma.
2.5 Réglage des options MCMC
L’onglet suivant, MCMC, fournit des réglages plus généraux pour contrôler la longueur de l’exécution MCMC et les noms de fichiers.
Premièrement, nous avons la longueur de la chaîne. C’est le nombre de pas que le MCMC fera dans la chaîne avant de se terminer. La longueur de la chaîne dépend de la taille de l’ensemble de données, de la complexité du modèle et de la qualité de la réponse requise. La valeur par défaut de 10 000 000 est entièrement arbitraire et doit être ajustée en fonction de la taille de votre ensemble de données. Pour cet ensemble de données, fixons la longueur de la chaîne à 6 000 000, car cela s’exécutera assez rapidement sur la plupart des ordinateurs modernes (quelques minutes).
Le champ Store Every détermine la fréquence à laquelle l’état est stocké dans le fichier. Stocker l’état périodiquement est utile pour les situations où l’environnement informatique n’est pas très fiable et où une exécution de BEAST peut être interrompue. Le fait de disposer d’une copie stockée de l’état récent vous permet de reprendre la chaîne au lieu de recommencer depuis le début, de sorte que vous n’avez pas besoin de repasser par le burn-in.Le champ Pre Burnin spécifie le nombre d’échantillons qui ne sont pas enregistrés au tout début de l’analyse. Nous laissons les champs Store Every et Pre Burnin sur leurs valeurs par défaut. En dessous de ces champs se trouvent les détails des fichiers journaux. Chacun peut être développé en cliquant sur le triangle noir.
Les options suivantes spécifient la fréquence à laquelle les valeurs des paramètres de la chaîne de Markov doivent être affichées à l’écran et enregistrées dans le fichier journal.La sortie écran sert simplement à contrôler la progression du programme et peut être réglée sur n’importe quelle valeur (bien que si elle est trop petite, la quantité d’informations affichées à l’écran ralentira le programme). Pour le fichier journal, la valeur doit être fixée par rapport à la longueur totale de la chaîne. Si vous échantillonnez trop souvent, vous obtiendrez des fichiers très volumineux avec peu d’avantages supplémentaires en termes de précision de l’analyse. Si l’échantillonnage est trop peu fréquent, le fichier journal n’enregistrera pas suffisamment d’informations sur les distributions des paramètres. Vous voulez probablement viser à stocker pas plus de 10 000 échantillons, donc cela devrait être fixé à pas moins de longueur de chaîne / 10 000.
Pour cet exercice, nous allons définir la fréquence du journal de trace et du journal d’arbre à 1 000 et le journal d’écran à 10 000.Spécifiez également Primates.log comme nom de fichier du fichier de journal de trace et Primates.trees comme nom de fichier du fichier de journal d’arbre.Assurez-vous que le nom de fichier déposé du journal d’écran est laissé vide,ou le journal d’écran ne sera pas écrit à l’écran.
- Si vous utilisez le système d’exploitation Windows, alors nous vous suggérons d’ajouter le suffixe .txt à ces deux fichiers (donc,Primates.log.txt et Primates.trees.txt) afin que Windows les reconnaisse comme des fichiers texte.
2.6 Générer le fichier XML BEAST
Nous sommes maintenant prêts à créer le fichier XML BEAST. Pour ce faire, sélectionnez l’option Enregistrer dans le menu Fichier. Vérifiez les prieurs par défaut, et enregistrez le fichier avec un nom approprié(nous terminons généralement le nom du fichier par .xml, c’est-à-dire Primates.xml).Nous sommes maintenant prêts à exécuter le fichier par BEAST.
3 Exécution de BEAST
Figure 12 : Une capture d’écran de BEAST.
Maintenant, exécutez BEAST et lorsqu’il demande un fichier d’entrée, fournissez votre fichier XML nouvellement créé comme entrée. BEAST fonctionnera alors jusqu’à ce qu’il ait fini de rapporter des informations à l’écran. Les fichiers de résultats réels sont enregistrés sur le disque au même endroit que votre fichier d’entrée. La sortie à l’écran ressemblera à quelque chose comme ceci :
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Notez qu’il y a quelques informations utiles au début concernant les alignements et les vraisemblances d’arbres utilisées. De plus, toutes les citations pertinentes pour l’analyse sont mentionnées au début de l’exécution, ce qui peut facilement être copié dans des manuscrits relatant l’analyse. Ensuite, suit le rapport de la chaîne, qui donne un certain feedback en temps réel sur la progression de la chaîne.
À la fin, une analyse des opérateurs est imprimée, qui liste tous les opérateurs utilisés dans l’analyse avec la fréquence à laquelle l’opérateur a été essayé, accepté et rejeté (voir les colonnes #total, #accept et #reject respectivement). Le taux d’acceptation est la proportion de fois où un opérateur est accepté lorsqu’il est sélectionné pour faire une proposition. En général, un taux d’acceptation élevé, disons supérieur à 0,5, indique que les propositions sont conservatrices et n’explorent pas efficacement l’espace des paramètres. D’autre part, un taux d’acceptation faible indique que les propositions sont trop agressives et aboutissent presque toujours à un état qui est rejeté en raison de sa faible postériorité.Les taux d’acceptation trop élevés et trop faibles entraînent tous deux des valeurs ESS faibles. Un taux d’acceptation de 0,234 est l’objectif (basé sur des preuves très limitées fournies par ) pour de nombreux opérateurs (mais pas tous) mis en œuvre dans BEAST.
Certains opérateurs ont un paramètre d’accord, par exemple le facteur d’échelle du paramètre ascale. Si le taux d’acceptation final n’est pas proche de la cible, BEAST suggère une nouvelle valeur pour le paramètre d’accord, qui est imprimée dans l’analyse de l’opérateur. Dans ce cas, tous les taux d’acceptation sont bons pour les opérateurs qui ont des paramètres de réglage. Les opérateurs sans paramètres de réglage comprennent les opérateurs wideexchange et Wilson-Balding pour cette analyse. Ces deux opérateurs tentent de changer la topologie de l’arbre avec de grands pas, mais comme les données soutiennent une topologie unique de manière écrasante, ces propositions radicales sont presque toujours rejetées.
4 Analyse des résultats
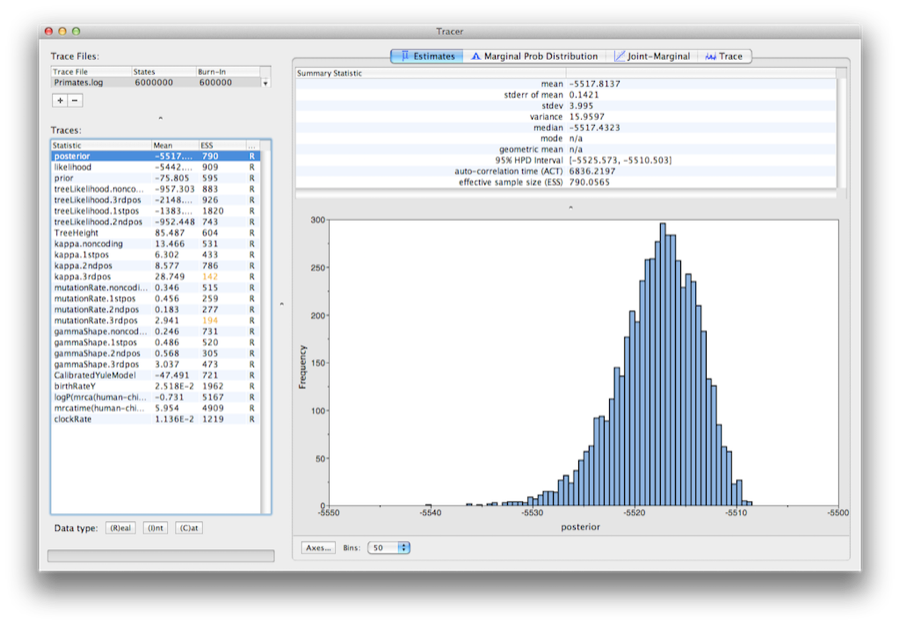
Figure 13 : Une capture d’écran de Tracer v1.6.
Lancez le programme appelé Tracer pour analyser la sortie de BEAST. Lorsque la fenêtre principale s’est ouverte, choisissez Importer un fichier de traces… dans le menu Fichier et sélectionnez le fichier que BEAST a créé et qui s’appelle Primates.log (figure 13).
N’oubliez pas que le MCMC est un algorithme stochastique et que les chiffres réels ne seront donc pas exactement les mêmes que ceux représentés sur la figure.
Sur le côté gauche se trouve une liste des différentes quantités que BEAST a enregistrées dans le fichier. Il y a des traces pour le postérieur (c’est le logarithme naturel du produit de la vraisemblance de l’arbre et de la densité antérieure), et les paramètres continus. La sélection d’une trace sur la gauche fait apparaître des analyses pour cette trace sur la droite, en fonction de l’onglet qui est sélectionné. Lors de la première ouverture, la trace « postérieure » est sélectionnée et diverses statistiques de cette trace sont affichées sous l’onglet Estimations.En haut à droite de la fenêtre se trouve un tableau des statistiques calculées pour la trace sélectionnée.
Sélectionnez le paramètre clockRate dans la liste de gauche pour regarder la vitesse moyenne d’évolution (moyenne sur tout l’arbre et tous les sites). Tracer tracera un histogramme (postérieur marginal) pour la statistique sélectionnée et vous donnera également des statistiques sommaires telles que la moyenne et la médiane. Le 95% HPD signifie intervalle de densité postérieure la plus élevée et représente l’intervalle le plus compact sur le paramètre sélectionné qui contient 95% de la probabilité postérieure. Il peut être considéré comme l’analogue bayésien d’un intervalle de confiance. Le paramètre TreeHeight donne la distribution postérieure marginale de l’âge de la racine de l’arbre entier.
Sélectionnez le paramètre TreeHeight, puis Ctrl-cliquez sur mrcatime(human-chimp) (Command-cliquez sur Mac OS X). Cela affichera l’âge de la racine et le MRCA de calibration que nous avons spécifié plus tôt dans BEAUti. Vous pouvez vérifier que la divergence que nous avons utilisée pour calibrer l’arbre(mrcatime(human-chimp)) a une distribution postérieure qui correspond à la distribution antérieure que nous avons spécifiée (Figure 14).
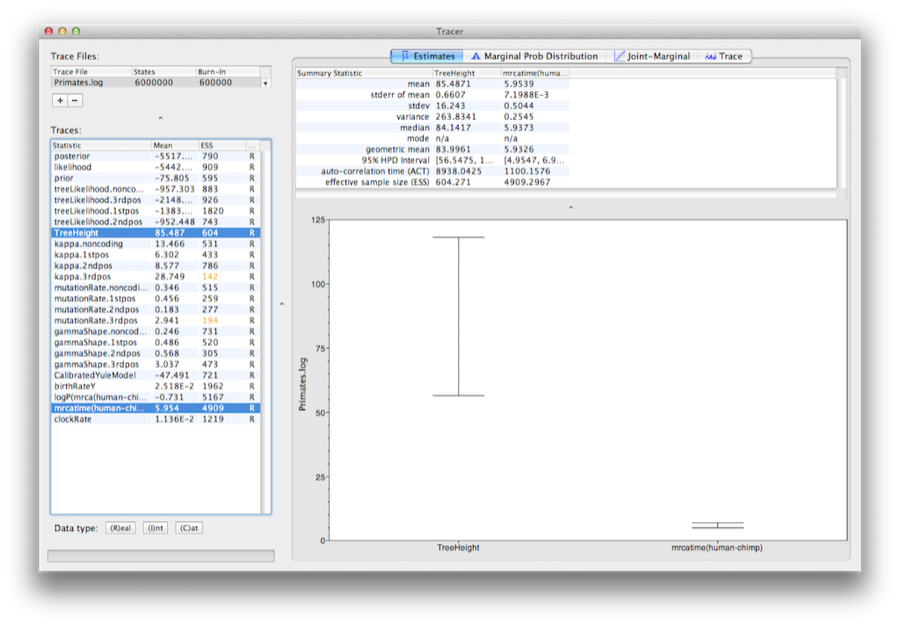
Figure 14 : Une capture d’écran des intervalles HPD à 95% de la hauteur de la racine et du MRCA (human-chimp) spécifié par l’utilisateur dans Tracer.
5 Estimations postérieures marginales
Pour montrer les taux relatifs pour les quatre partitions, sélectionnez le paramètre mutationRate pour chacune des quatre partitions, et sélectionnez l’onglet densité marginale dans Tracer.La figure 15 montre les densités marginales pour les taux de substitution relatifs. La figure 15 montre les densités marginales pour les taux de substitution relatifs. Le graphique montre que les positions de codon 1 et 2 ont des taux sensiblement différents (0,456 contre 0,183) et qu’elles sont toutes deux beaucoup plus lentes que la position de codon 3 avec un taux relatif de 2,941. La partition non codante a un taux intermédiaire entre les positions de codon 1 et 2 (0,346). Pris ensemble, ce résultat suggère une forte sélection purificatrice dans les régions codantes et non codantes de l’alignement.
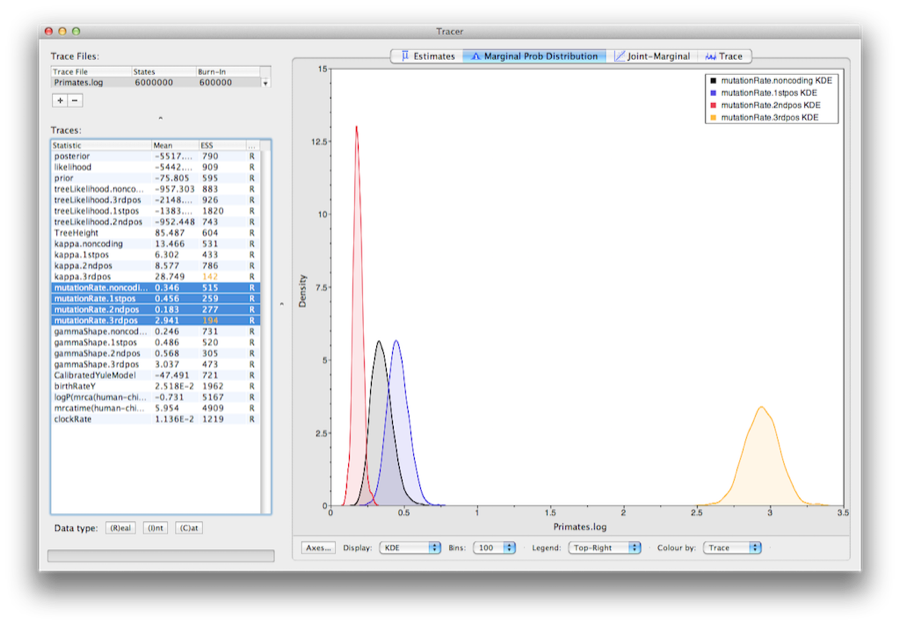
Figure 15 : Une capture d’écran des densités marginales postérieures des taux de substitution relatifs des quatre partitions (par rapport au taux moyen pondéré par site).
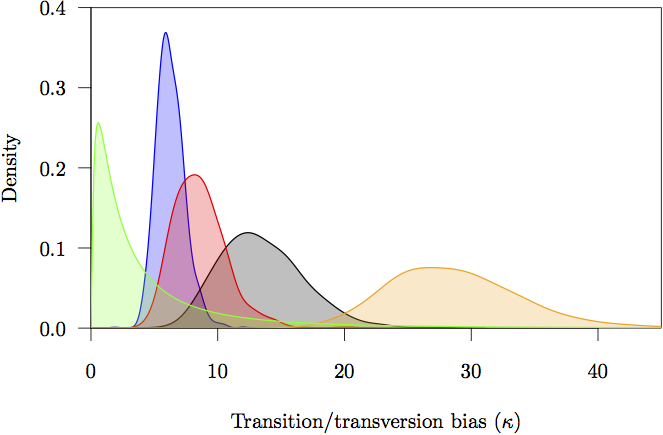
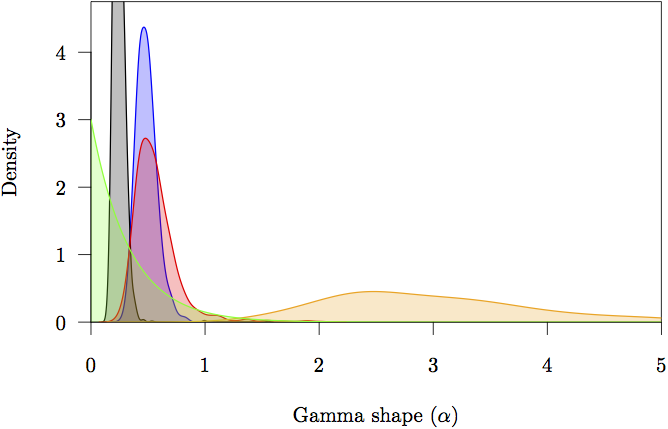
Figure 16 : Les densités marginales antérieures et postérieures pour les paramètres de forme (α). L’antériorité est en gris. L’estimation de la densité postérieure pour chaque partition est également montrée : non codant (orange) et première (rouge), deuxième (verte) et troisième (bleue) positions de codon.
Figure 17 : Les densités marginales antérieures et postérieures pour les paramètres de biais de transition/transversion (κ). L’antériorité est en gris. L’estimation de la densité postérieure pour chaque partition est également montrée : positions non codantes (orange) et première (rouge), deuxième (verte) et troisième (bleue) positions de codon.
Questions
Quel est le taux estimé d’évolution moléculaire pour cet arbre génique (inclure l’intervalle HPD de 95%) ?
Quelles sources d’erreur cette estimation inclut-elle ?
Quel est l’âge de la racine de l’arbre (donnez la moyenne et l’intervalle HPD de 95%) ?
6 Obtention d’une estimation de l’arbre phylogénétique
BEAST produit également un échantillon postérieur d’arbres temporels phylogénétiques avec son échantillon d’estimations de paramètres. Ceux-ci doivent être résumés à l’aide du programme TreeAnnotator. Celui-ci prendra l’ensemble des arbres et trouvera celui qui est le mieux supporté. Il annotera ensuite cet arbre récapitulatif représentatif avec les âges moyens de tous les nœuds et les plages de DHP à 95 % correspondantes. Il calculera également la probabilité postérieure des clades pour chaque nœud. Exécutez le programme TreeAnnotator et configurez-le comme indiqué sur la figure 18.
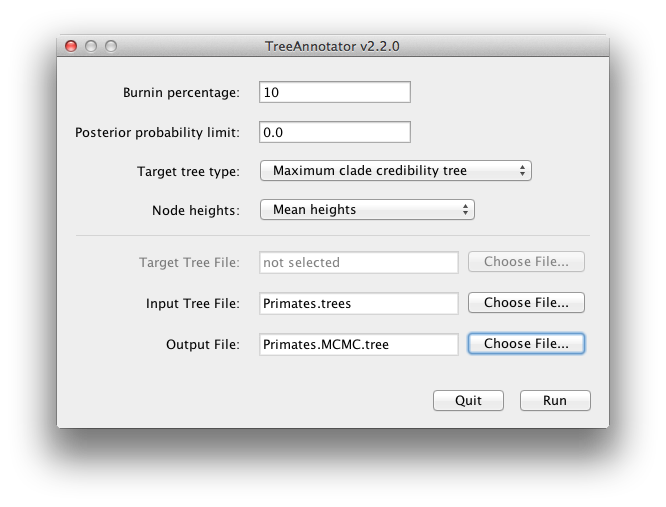
Figure 18 : Une capture d’écran de TreeAnnotator.
Le burnin est le nombre d’arbres à retirer du début de l’échantillon. Contrairement à Tracer qui spécifie le nombre de pas comme burnin, dans TreeAnnotator vous devez spécifier le nombre réel d’arbres. Pour cette exécution, vous avez spécifié une longueur de chaîne de 6 000 000 de pas, avec un échantillonnage tous les 1 000 pas. Ainsi, le fichier d’arbres contiendra 6 000 arbres et donc de spécifier un burnin de 10% dans le champ de texte supérieur.
L’option de limite de probabilité postérieure spécifie une limite telle que si un nœud est trouvé à moins de cette fréquence dans l’échantillond’arbres (c’est-à-dire, a une probabilité postérieure inférieure à cette limite), il ne sera pas annoté. La valeur par défaut de 0,5 signifie que seuls les nœuds vus dans la majorité des arbres seront annotés. Définissez cette valeur à zéro pour annoter tous les nœuds.
Le type d’arbre cible spécifie la topologie de l’arbre qui sera annotée. Vous pouvez soit choisir un arbre spécifique à partir d’un fichier, soit demander à TreeAnnotator de trouver un arbre dans votre échantillon.L’option par défaut, l’arbre de crédibilité de clade maximale, trouve l’arbre avec le produit le plus élevé de la probabilité postérieure de tous ses nœuds.
Pour les hauteurs de nœud, la valeur par défaut est Common Ancestor Heights, qui calcule la hauteur d’un nœud comme la moyenne du temps MRCA de toutes les paires de nœuds dans le clade. Pour les arbres dont la topologie est très incertaine et qui présentent donc de nombreux clades à faible soutien, certaines autres méthodes peuvent donner des arbres à longueur de branche négative. Dans cette analyse, le support de tous les clades dans l’arbre résumé est très élevé, donc ce n’est pas un problème ici.Choisissez Hauteurs moyennes pour les hauteurs des nœuds. Cela définit les hauteurs (âges) de chaque nœud de l’arbre à la hauteur moyenne à travers l’échantillon entier d’arbres pour ce clade.
Pour le fichier d’entrée, sélectionnez le fichier d’arbres que BEAST a créé et sélectionnez un fichier pour la sortie (ici nous l’avons appelé Primates.MCC.tree). Appuyez maintenant sur Run et attendez que le programme se termine.
7 Visualisation de l’estimation de l’arbre
Enfin, nous pouvons visualiser l’arbre dans un autre programme appelé FigTree. Exécutez ce programme, et ouvrez le fichier Primates.MCC.tree en utilisant la commande Open du menu File. L’arbre devrait apparaître. Vous pouvez maintenant essayer de sélectionner certaines des options dans le panneau de contrôle sur la gauche. Tout d’abord, étendez l’option Trees dans le panneau, et cochez Order nodes et choisissez Ordering by decreasing. Essayez de sélectionner Node Bars pour obtenir les barres d’erreur de l’âge des nœuds. Activez également l’option Branch Labels et sélectionnez posterior pour que l’outil affiche la probabilité postérieure de chaque nœud. Si vous utilisez un modèle d’horloge non strict, alors sous Apparence, vous pouvez également dire à FigTree de colorer les branches par le taux.Vous devriez vous retrouver avec quelque chose de similaire à la figure 19.
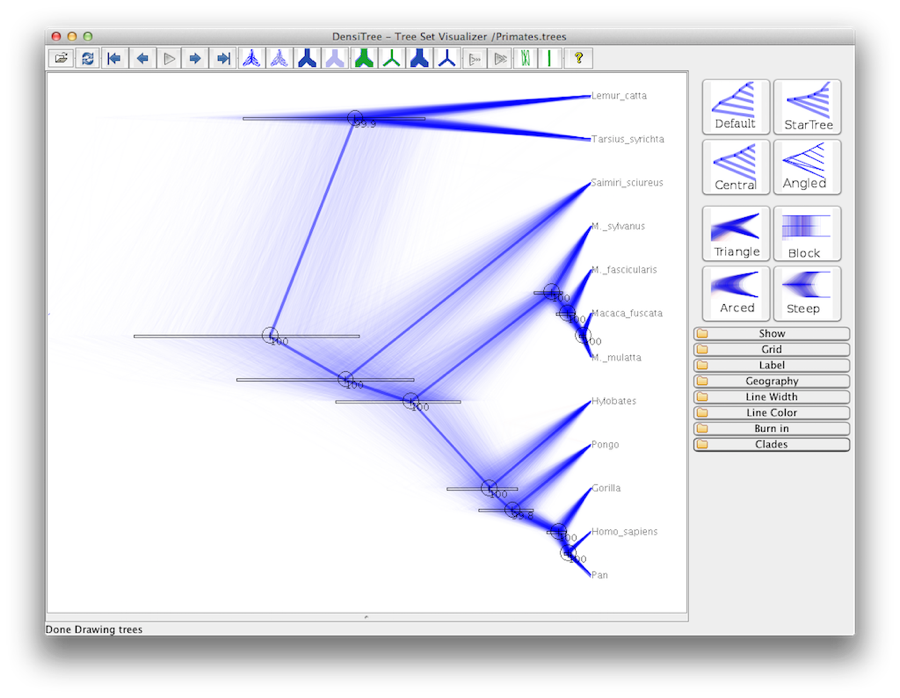
Figure 19 : Une capture d’écran de FigTree et DensiTree.
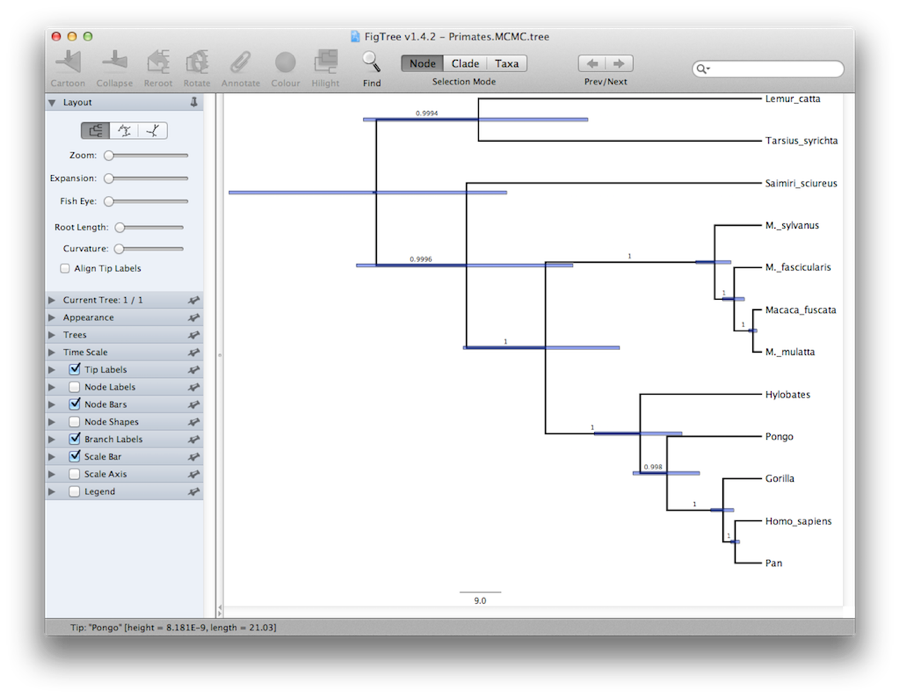
Une vue alternative de l’arbre peut être faite avec DensiTree, qui fait partie de Beast 2. L’avantage de DensiTree est qu’il est capable de visualiser à la fois l’incertitude dans les hauteurs des nœuds et l’incertitude dans la topologie.Pour ce jeu de données particulier, la topologie dominante est présente dans plus de 99% des échantillons. Nous concluons donc que cette analyse aboutit à un consensus très élevé sur la topologie (figure 19).
Questions
- Le taux d’évolution diffère-t-il sensiblement parmi les différentes lignées de l’arbre ?
- DensiTree possède une barre de clades (Menu Window/View clade toolbar) pour afficher des informations sur les clades.
Quel est le support du clade ?
- Vous pouvez parcourir les topologies dans DensiTree en utilisant le menu Browse.La topologie la plus populaire a un support de plus de 99%.
Quel est le support de la deuxième topologie la plus populaire ?
- Dans le menu d’aide, DensiTree affiche quelques informations.
Combien de topologies sont dans l’ensemble d’arbres ?
8 Comparer vos résultats à l’antériorité
C’est une bonne idée de réexécuter l’analyse tout en échantillonnant à partir de l’antériorité pour s’assurer que les interactions entre les antériorités n’affectent pas votre information antérieure. L’interaction entre les prieurs peut être problématique, en particulier lors de l’utilisation de calibrations, car cela signifie mettre plusieurs prieurs sur l’arbre.
En utilisant BEAUti, configurez la même analyse, mais sous les options MCMC, sélectionnez l’option Echantillonnage à partir du préalable uniquement. Cela vous permettra de visualiser la distribution complète des priors en l’absence de vos données de séquence. Résumez les arbres à partir de la distribution complète des antériorités et comparez le résumé à l’arbre sommaire postérieur.
L’estimation du temps de divergence à l’aide de la « datation des nœuds » du type décrit dans cechapitre a été appliquée pour répondre à une variété de questions différentes en écologieet en évolution. Par exemple, la datation des nœuds à l’aide de fossiles a été utilisée pour déterminer la diversité des espèces de cycadés, analyser le taux d’évolution des plantes à fleurs et étudier les origines des cyanobactéries des déserts chauds et froids.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond et Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), no. 1, 114. A Gelman, G Roberts, et W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled et Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation,Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, et S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian, and Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith et Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008),no. 5898, 86-89.
Ce document a été traduit de LATEX parHEVEA.