John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Az egyre több bizonyíték világít rá a II. típusú cukorbetegség (T2D) és az Alzheimer-kór (AD) közötti szoros kapcsolatra. Fontos, hogy e két betegség számos patológiai hasonlóságot mutat, beleértve az amiloid felhalmozódást, az oxidatív stresszt, a gyulladást és a sejthalált. A mai napig nincsenek gyógyszeres terápiák az Alzheimer-kór és a T2D kezelésére, és alapvető szükség van az új terápiák felfedezésére és fejlesztésére ezekre a betegségekre. Számos humán és rágcsálókon végzett vizsgálat bizonyította, hogy a metabolikus hormonpótlás rendkívül értékes a kognitív funkciók és az általános metabolikus egészség javítása szempontjából mind a T2D, mind az AD esetében. A hasnyálmirigy-hormon, az amilin mind a T2D, mind az Alzheimer-kór etiológiájának kulcsfontosságú összetevőjeként jelent meg, bár az amilin pontos szerepe ezekben a betegségekben még nem teljesen ismert. Itt kritikusan áttekintjük a jelenlegi irodalmat, amely a humán amilint vagy annak szintetikus analógját, a pramlintidot, valamint az amilinreceptor-antagonistákat használja az AD kezelésére.
Bevezetés
Az Alzheimer-kór (AD) progresszív, legyengítő neurodegeneratív betegség, amelyet az amiloid-béta (Aβ) plakkok és a hiperfoszforilált tau1-ből álló neurofibrilláris csomók felhalmozódása jellemez. E kóros peptidek felhalmozódása hozzájárul a végrehajtó funkciók, például a tanulás és a memória, a hangulat, az érzelmek stb. hiányosságaihoz, és jelentős terhet jelent a beteg és a gondozók számára. Az Alzheimer-kór előfordulása riasztó mértékben növekszik az Egyesült Államokban: 2017-ben becslések szerint 5,5 millió amerikai élt Alzheimer-kórral, és ez a szám 2050-re várhatóan megháromszorozódik2. Ezenkívül az Alzheimer-kóros betegek gondozásának és kezelésének költségei jelenleg meghaladják az évi 200 milliárd dollárt, és várhatóan csak növekedni fognak3. Bár az Alzheimer-kór egyértelműen monumentális problémát jelent az Egyesült Államokban és azon kívül is, a kezelési lehetőségek továbbra is nagyon korlátozottak4. Számos gyógyszerkísérletet végeztek célzott megközelítések széles skálájával, mégis jelenleg csak hat gyógyszert engedélyezett az FDA az Alzheimer-kór kezelésére, és ezek csak tüneti kezelések5,6. Eddig a kifejlesztett farmakológiai szerek többsége kifejezetten az Aβ vagy a tau patológiáját célozta meg, de egyik sem volt sikeres a patológia megszüntetésében vagy megelőzésében4. Ezért alapvető szükség van az Alzheimer-kór életképes terápiás és megelőző kezeléseinek kifejlesztésére.
Az életkorral összefüggő (sporadikus) Alzheimer-kór egy bonyolult, multifaktoriális betegség, amelynek számos genetikai és környezeti hatása van. A környezet és az életmód jelentős szerepet játszik a sporadikus Alzheimer-kór kialakulásában; olyan tényezők, mint az étrend7-9, az elhízás8-10, a metabolikus szindróma7, a II. típusú cukorbetegség (T2D)9, 11 és a szív- és érrendszeri betegségek12 mind szerepet játszanak az Alzheimer-kór kialakulásában. Kritikus jelentőségű, hogy az elhízás és a cukorbetegség aránya az Alzheimer-kórral párhuzamosan gyorsan emelkedik12, 13. Bár az elhízás és az Alzheimer-kór közötti kapcsolat némileg tisztázatlan, bizonyíték van arra, hogy a középkorú elhízás szerepet játszik az Alzheimer-kór kialakulásában10. Ami még fontosabb, hogy az elhízást általában számos más betegség is kíséri, beleértve a szív- és érrendszeri betegségeket, a magas vérnyomást, a diszlipidémiát, a T2D-t, a stroke-ot stb.14. A T2D előfordulása gyorsan növekszik, a CDC becslése szerint az USA-ban körülbelül 30,3 millió ember (10 felnőttből 1) cukorbeteg, és megdöbbentő 84,1 millió (3 felnőttből 1) prediabéteszes, akiknek többsége nem tud állapotáról. Továbbá, a fizikai aktivitás nagymértékű csökkenése miatt, amelyet a táplálékbevitel és a helytelen táplálkozás egyidejű növekedése kísér, az elhízás, a T2D, a metabolikus szindróma és a szív- és érrendszeri betegségek aránya a becslések szerint 2035-re világszerte 600 millió T2D-esetre fog emelkedni15.
Az anyagcsere-funkciót és a betegségeket a kognitív hanyatlás és az öregedés folyamatában szerepet játszó bizonyítékok száma jelentős16, 17 . Például a T2D-vel diagnosztizált emberek mintegy 70%-a számol be kognitív károsodásról, és a T2D-s betegek jelentős részénél később demencia alakul ki16, 18-21. A legalább öt éve T2D-vel diagnosztizált személyeknél jelentősen megnövekedett az Alzheimer-kór kialakulásának kockázata azokhoz képest, akik kevesebb mint öt éve szenvednek T2D-ben17. Ezek az adatok együttesen arra utalnak, hogy a T2D növekvő előfordulása a lakosság körében hozzájárulhat az Alzheimer-kór növekvő arányához.
A T2D-t kezdetben magas vércukorszint és inzulinszint jellemzi, ami hiperinsulinaemiához vezet; fontos, hogy az amilin, egy kis metabolikus hormon, amelyet a hasnyálmirigy β-szigetsejtjei termelnek, az inzulinnal együtt csomagolódik és együtt szekretálódik, így T2D-ben túltermelődik22. Fontos, hogy számos olyan patológiai jellemző van, amely mind a T2D-ben, mind az Alzheimer-kórban jelen van: 1) csökkent agyi anyagcsere és metabolikus hormonrezisztencia 2) amiloid patológia 3) oxidatív stressz (OS) és gyulladás. A krónikus, hiperinsulinémia és hiperamylinémia számos fiziológiai problémához vezet: a krónikus hiperinsulinémia a rendszer inzulinrezisztenciájához22, a vér-agy gáton (BBB)23, 24 keresztül történő inzulintranszport károsodásához, és így az agyon belüli inzulinszignalizáció csökkenéséhez vezet25. Az inzulinszignalizáció elvesztése az agyban számos AD-hez kapcsolódó patológiai jellemzővel jár együtt, beleértve a fokozott Aβ-termelődést, a tau-foszforilációt és a neuroinflammációt.
Az amilin továbbá magas koncentrációban hasonló patológiai jellemzőkkel rendelkezik, mint az Aβ26 , és közös útvonalat jelenthet a két betegség között. Az amilin fibrillákat például a T2D-s betegek 95%-ának hasnyálmirigyében találták27-29 , és számos élettani zavart okoznak, beleértve az aberráns Ca2+ beáramlást, a pro-inflammatorikus citokinek fokozott szekrécióját30,31 és végül a β-szigetsejtek elvesztését32. Ezenkívül az amilin könnyen átjut a BBB-n és amilin fibrillákat, valamint Aβ-val kevert plakkokat képez az agyban, és felelős lehet az AD-szerű patológiáért és az Aβ elszaporodásáért a T2D-ben33-35 . Ismert, hogy az amilin befolyásolja a hosszú távú potenciálódást (LTP) a hippokampuszban, és veleszületett hatással lehet az agyon belüli kognitív funkciókra36-39. Az azonban, hogy az amylin toxikus inzultus-e ezekben a betegségekben, vagy hogy funkcionális elvesztése aggregáció vagy késői stádiumú β-sejtvesztés révén T2D-ben hozzájárul-e az AD kialakulásához, továbbra is tisztázatlan.
Az amylin-szignalizációs dichotómia
Még mindig sok vita folyik az amylin-receptor (AMYR) és az amylin-szignalizáció részvételéről a T2D és az AD betegségprogressziójában és etiológiájában. Az ennek a kapcsolatnak a felismerését célzó kutatások tömege gyorsan bővül. Minden releváns kutatás következetesen kimutatta, hogy az amilin jelátvitel modulációja befolyásolja az AD-vel kapcsolatos patológiát. Ennek a kapcsolatnak a természetét azonban még nem sikerült konkrétan tisztázni. Több csoport is meggyőző adatokat szolgáltatott, amelyek arra utalnak, hogy az amilin jelátvitel mind in vivo, mind in vitro előnyös az Alzheimer-kórhoz kapcsolódó patológia és a kognitív deficitek megelőzésében40-44 . Fontos, hogy a pramlintid, az amilin rekombináns, nem aggregáló formája, amelyet inzulinterápiákkal együtt alkalmaznak a cukorbetegség kezelésére, és amely javítja a glikémiás kontrollt, csökkenti a testsúlyt és csökkenti az OS szérummarkereit45-47 , szintén ígéretesnek tűnik AD-terápiaként. A mai napig azonban nem végeztek olyan klinikai vizsgálatokat, amelyek az amilin vagy a pramlintid terápiás szerként való felhasználását célozták volna a demencia kezelésében. Rágcsálókon végzett vizsgálatokból származó egyértelmű bizonyítékok arra utalnak, hogy a humán amilinnel vagy pramlintiddel végzett krónikus kezelés erős terápiás előnyt jelent az Alzheimer-kórhoz kapcsolódó patológia csökkentésében; az amilin/pramlintid kiegészítés csökkenti az oldható Aβ-szintet, a plakkterhelést, a tau-foszforilációt, a neuroinflammációt és az OS-t, miközben a kogníciót is javítja40-42,44 . A fenti adatok arra utalnak, hogy a veleszületett amylin-szignalizáció elvesztése a CNS-ben az aggregáció miatt fokozott kockázatot jelent az AD kialakulására, és ezzel részletesebben foglalkozik Grizzanti és munkatársai 201848.
A vizsgálatok ezzel szemben azt is mutatják, hogy a humán amylin és az Aβ hasonló toxikus hatással rendelkezik, és hogy ezek a toxikus hatások enyhíthetők AMYR-antagonistával36-39,49. Az adatok például azt mutatják, hogy az AMYR-antagonistákkal végzett in vivo kezelés nagyon hasonló élettani előnyöket eredményez, mint az amilin- vagy pramlintidkezelés. A TgCRND8 AD egerek kezelése AC253-mal, egy AMYR-antagonistával vagy ciklikus megfelelőjével, a cAC253-mal csökkenti a neuroinflammációt, az oldható Aβ-szintet és a plakkterhelést, miközben a kogníciót is javítja50. Hasonlóképpen, in vitro/ex-vivo vizsgálatok azt mutatják, hogy az alacsony dózisú humán amylin vagy Aβ zavarokat okoz az LTP-ben, és hogy ezeket a hiányosságokat az AC253 vagy a pramlintid blokkolja38,39, és a humán amylin/amylin oligomerek nagyobb dózisai kontrollálatlan Ca2+ beáramlással járnak, ami szorosan összefügg a sejthalállal26,32. Ezek az adatok együttesen alátámasztják az amilin oligomerek toxikus funkcióját, és így az AMYR-blokád potenciális terápiás mechanizmusát. Ezzel szemben mások kimutatták, hogy az amylin kedvező hatásai az AC25341 segítségével blokkolhatók. Így az amilin kezelés vagy gátlás terápiás potenciálja továbbra is tisztázatlan, és rávilágít az amiloidok komplex és dichotóm természetére az agyban és a periférián.
Piecing Together the Puzzle
A jelenlegi irodalomban számos lyuk van, amelyeket be kell tölteni ahhoz, hogy teljesebb képet kapjunk az amilin történetéről: 1) a veleszületett amilin rendszer és az agyon belüli amilin jelátvitel természete 2) az Aβ és pramlintid jelátviteli képességek a három fő AMYR és rokon receptoron keresztül 3) az amilin/pramlintid vagy AMYR gátlás közvetítésének terápiás mechanizmusai. Először is, érdekes új adatok bizonyítják, hogy az AMYR nemcsak a jelátvitelben, hanem a BBB-n keresztüli ligandszállításban is részt vesz. Az AMYR egy heterodimer receptor, amely egy kalcitoninreceptorból és egy receptoraktivitást módosító fehérjéből (1-3)51 áll. Ennek érdekében a kalcitoninreceptor (az AMYR egyik kulcskomponense) 50%-os globális knockdownja jelentősen csökkentette az agyban található AC253 mennyiségét50 , ami azt jelzi, hogy a BBB-ben található AMYR részt vesz e ligandumok agyba történő szállításában, és az amilin és a pramlintid agyba/agyból történő ki/be szállításában is szerepet játszhat. E BBB-transzportmechanizmusok létezése arra utal, hogy az amilin valószínűleg veleszületett fiziológiai funkcióval rendelkezik az agyban, mivel az agyba történő szállítása szigorúan szabályozott. Az azonban, hogy az amilin jelátvitel vagy annak hiánya hogyan vezet az Alzheimer-kór patológiás jellemzőihez, és hogy az AMYR-e az a jármű, amelyen keresztül az Aβ közvetíti toxikus hatásait, még mindig tisztázatlan.
Az Aβ és az AMYR közötti kapcsolatra vonatkozóan ellentmondásos bizonyítékok állnak rendelkezésre. Bár számos vizsgálat egyértelműen bizonyítja, hogy a humán amylin és az Aβ hasonló hatással van a CNS-ben az LTP-re, és az AMYR-gátlók alkalmazása javítja ezeket a káros hatásokat36-39, más bizonyítékok arra utalnak, hogy az Aβ (1-42) képtelen az AMYR bármelyikén keresztül jelezni, hogy bármilyen cAMP-választ kiváltson a legkülönbözőbb koncentrációkban52. Lehetséges, hogy az Aβ az AMYR-rel való kölcsönhatás révén különböző jelátviteli kaszkádokat aktivál, vagy egyszerűen inert kompetitív gátlóként viselkedik, de ezt még nem sikerült bizonyítani.
Egy külön vizsgálatban kimutatták továbbá, hogy az oligomer amilin közvetlenül az AMYR-en keresztül, közvetve pedig a TRPV4-en, egy nem szelektív kationcsatornán keresztül közvetíti toxikus hatásait26. A humán amilin alacsony koncentrációi olyan Ca2+ választ váltanak ki, amelyet a natív receptor közvetít. Magasabb koncentrációban azonban a humán amilin oligomereket képez és aberrált jelátvitelt vált ki, ami a TRVP4 csatornák aktiválódását eredményezi, és lehetővé teszi a kontrollálatlan kationbeáramlást, különösen a Ca2+ -ot. Az AMYR és a TRPV4 farmakológiai blokkolása azt mutatja, hogy mindkét receptor szükséges ahhoz, hogy az oligomer humán amilin kiváltsa toxikus Ca2+ hatását26. Így valószínű, hogy az Aβ hasonló módon közvetíti toxikus hatásait az AMYR-en, bár ezek az adatok még nem állnak rendelkezésre. A kontrollálatlan Ca2+-beáramlás számos patológiás jelenséghez kapcsolódik, beleértve a kontrollálatlan vesikuláris felszabadulást, az OS és a mitokondriumok diszfunkcióját, az apoptózist stb. Ennek érdekében valószínű, hogy a toxikus amiloid jelátvitelből eredő sejtszintű diszfunkció és a további AD-szerű patológia kialakulása mind az AMYR-en, mind a TRPV4-en keresztül közvetít. Mint ilyen, meg kell különböztetni azokat a jelátviteli kaszkádokat, amelyek modulálják az AMYR és a TRVP4 közötti kapcsolatot. Továbbá indokoltak olyan farmakológiai kísérletek, amelyek az Aβ és a pramlintid széles dózistartományban használják az Aβ és a pramlintid Ca2+ áramokra, LTP-re, cAMP-termelésre és más jelátviteli kaszkádokra gyakorolt hatását, hogy meghatározzák jelátviteli képességeiket. Ezek a kísérletek segítenek betölteni a jelenlegi irodalomban az AMYR-rel és annak a betegségállapotokban való részvételével kapcsolatos néhány űrt (1. ábra).
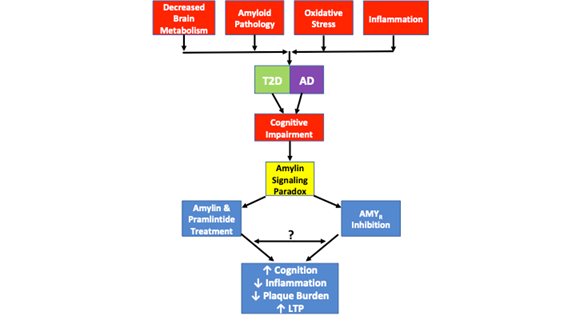
Az 1. ábra az amylin jelátviteli paradoxont és a T2D-ben és az AD-ben megfigyelt patológiai hasonlóságokat ábrázolja. A csökkent agyi anyagcsere, az amiloid patológia, az oxidatív stressz és a gyulladás mind közös patológiai jellemzők, amelyek mindkét betegségben megfigyelhetők. Bár a T2D vagy az AD nem minden esete tartalmazza e patológiai jellemzők mindegyikét, minden esetben megfigyelhető a kognitív károsodás. Az amilin jelátviteli paradoxon a képbe kerül, mivel vizsgálatok kimutatták, hogy mind az AMYR gátlása, mind az AMYR agonizmus az amilin és a pramlintid kezelésen keresztül a kogníció javulását, a gyulladás csökkenését, a plakkterhelés csökkenését és az LTP növekedését eredményezi. Az AMYR-agonizmus és az AMYR-antagonizmus által szabályozott jelátviteli mechanizmusok még nem teljesen tisztázottak. Míg az amylin jelátvitelt hagyományosan a cAMP és a PKA jelátvitellel hozzák összefüggésbe, nem világos, hogy más kaszkádokat is aktivál-e az amylin/pramlintid. Továbbá nem világos, hogy az AMYR-antagonisták, az amilin oligomerek vagy az Aβ az AMYR-en keresztül jeleznek-e, illetve hogy van-e hasonlóság vagy keresztbeszélgetés mindezen AMYR-ligandumok között. Mint ilyen, az ebben az áttekintésben javasolt számos kísérlet segíteni fog az AMYR valódi természetének további tisztázásában.
Következtetések
Az amilin jelátvitel agyban betöltött szerepével kapcsolatos jelenlegi ellentmondás azt mutatja, hogy alapvető szükség van az amilin szerepének további tisztázására mind az Alzheimer-kórban, mind a T2D-ben. A T2D-ben valószínű, hogy a betegség korai szakaszában az amilin elárasztja az agyat, oligomereket képez, aberráns jelátvitelt indukál natív receptorán keresztül, és toborozza a TRPV4-et, hogy kóros Ca2+ beáramlást indukáljon, ami széles körű neuronális diszfunkciót eredményez, ami OS, kontrollálatlan vesikuláris felszabadulás és interneuronális diszfunkció, gyulladás és az ebből eredő sejthalál formájában jelentkezik. Ez a mechanizmus lehet felelős a kezdeti átmenetért az egészséges agyból az agyi öregedésbe a metabolikus betegségekben. Mint ilyen, az AMYR vagy a TRVP4 gátlása bizonyos időpontokban a metabolikus betegségekben és a cukorbetegség korai szakaszában indokolt lehet az oligomer amylin vagy az Aβ toxikus hatásainak blokkolása érdekében. Erős bizonyítékok utalnak azonban arra is, hogy az amilin helyettesítése humán amilinnel vagy pramlintiddel csökkenti az Alzheimer-kórhoz kapcsolódó főbb patológiák többségét, miközben javítja a kogníciót is az Alzheimer-kór rágcsálómodellekben. Ezért indokolt lehet az amilin jelátvitel helyettesítése amilinnel vagy pramlintiddel a cukorbetegség középső és késői szakaszában, amikor az amilin jelátvitel az aggregáció, oligomerizáció vagy a β-sejtek elvesztése miatt megszűnik. Ebből a célból szükség van a metabolikusan összefüggő agyi öregedés kóros eseményeinek időbeli megjelenésének és a korai, középső és késői stádiumú betegség terápiás lehetőségeinek megkülönböztetésére is. Ezen amiloidok közvetlen természetének és jelátviteli képességeinek, valamint a specifikus időbeli kezelések terápiás jellegének kritikus elemzése és vizsgálata segíthet áthidalni az AMYR-gátló terápiák és az amilint helyettesítő terápiák közötti szakadékot.
Finanszírozás
A cikk finanszírozását a National Institutes of Aging grant 1R15AG050292-01A1 biztosította.
- LaFerla FM, Oddo S. Alzheimer-kór: Aβ, tau és szinaptikus diszfunkció. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurológia. 2013; 80: 1778-1783.
- Association As. 2016 Alzheimer-kór tények és számok. Alzheimer-kór & Demencia. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer-kór gyógyszerfejlesztés pipeline: kevés jelölt, gyakori kudarcok. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glükózszint és a demencia kockázata. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of dementia prevalence in the United States and China. Obesity. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. A 2-es típusú cukorbetegség mint az Alzheimer-kór kockázati tényezője: az összefüggéssel kapcsolatos zavaró tényezők, kölcsönhatások és neuropathológia. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Az elhízás és társbetegségei. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF diabétesz atlasz. Brüsszel: Nemzetközi Diabétesz Szövetség. 2013.
- K Dash S. Kognitív károsodás és cukorbetegség. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. The lancet Diabetes & endokrinológia. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalence of diabetes and prediabetes and their risk factors among Bangladeshi adults: a nationwide survey. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Az inzulint lebontó enzim szabályozza az inzulin, az amiloid β-protein és a β-amyloid prekurzor protein intracelluláris doménjének szintjét in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptidek. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Az inzulinszintek csökkennek a prodomális Alzheimer-kórban szenvedő nők agy-gerincvelői folyadékában. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neurofarmakológia. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Az elhízást az inzulinrezisztenciával és a 2-es típusú cukorbetegséggel összekötő mechanizmusok. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. The American journal of pathology. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Az NLRP3 inflammaszóma aktiválása a sziget amiloid polipeptid által a 2-es típusú cukorbetegségben a fokozott IL-1β mechanizmusát biztosítja. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimer β-amyloid, humán szigeti amylin és prion fehérje fragmentum közös mechanizmus révén idéz elő intracelluláris szabad kalcium emelkedést egy hipotalamikus GnRH neuronális sejtvonalban. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. The American journal of pathology. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylin receptor: közös patofiziológiai célpont az Alzheimer-kórban és a cukorbetegségben. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ) peptid közvetlenül aktiválja az amylin-3 receptor altípust több intracelluláris jelátviteli útvonal kiváltásával. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide Antagonizes Beta Amyloid (Aβ)-and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloid-indukált depressziója a hippokampális hosszú távú potenciációnak az amilin receptoron keresztül közvetített. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. A hasnyálmirigy peptid amylin intraperitoneális injekciója hatékonyan csökkenti a viselkedési károsodást és az agyi amiloid patológiát az Alzheimer-kór egérmodelljében. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylin receptor ligandok csökkentik az Alzheimer-kór patológiai kaszkádját. Neuropharmacology. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Az amilin analóg pramlintid neuroprotektív hatása az Alzheimer-kór patogenezisére és kogníciójára. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. A β-katenin destabilizálása a presenilin-1 mutációi által potenciálja a neuronális apoptózist. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintid-acetát injekció az 1-es és 2-es típusú diabetes mellitus kezelésére. Clin Ther. 2007; 29: 535-562.
- Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta?analysis. Diabetes, Obesity and Metabolism. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Az amilin analógok neuroprotektív hatása az Alzheimer-kór patogenezisére és kogníciójára. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. A β-amyloid fehérje humán neuronokra gyakorolt hatása az amilin receptoron keresztül fejeződik ki. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyclic AC253, egy új amilin receptor antagonista, javítja a kognitív deficitet az Alzheimer-kór egérmodelljében. Alzheimer-kór & Demencia: Transzlációs kutatás & Klinikai beavatkozások. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. A pramlintid, patkány és humán amilin, de nem az Aβ1-42 aktivitása a humán amilin receptorokon. Endokrinológia. 2014; 155: 21-26.