Introduction
Germline de novo mutations (DNM) are genetic changes in the individual caused by mutagenesis occurring in parental gametes during oogenesis and spermatogenesis. Itt a “de novo” kifejezés nem tévesztendő össze az “új mutáció” kifejezéssel. Annak ellenére, hogy a DNM-ek egy trió (apa, anya és gyermek) kontextusában újszerű mutációk, lehetnek gyakori, ritka vagy újszerű változatok az általános populációban. Egy adott DNM arányának méréséhez és magyarázatához először a variáns fenotípusra gyakorolt hatását kell értékelni, mivel új kedvező tulajdonságok fejlődhetnek ki, ha a felmerülő genetikai mutációk konkrét túlélési előnyt kínálnak (Front Line Genomics, 2017).
A szórványosan előforduló, nem mendeli genetikai betegségekben szenvedő emberekben a DNM-ek általában újszerűek, megbízhatóbbak és károsabbak, mint az öröklött variánsok, mivel nincsenek kitéve erős természetes szelekciónak (Crow, 2000; Front Line Genomics, 2017). Ezért az egyénben egy DNM által kiváltott rendellenesség genetikai okának azonosítása klinikai szempontból kihívást jelenthet, mivel pleiotrópia és genetikai heterogenitás állhat egyetlen fenotípus hátterében (Eyre-Walker és Keightley, 2007). Ennek megfelelően az elmúlt évtizedben jelentős erőfeszítéseket tettek a nem egyértelmű genetikai etiológiájú betegségekben szenvedő egyének exomjának szekvenálására a klinikai diagnosztika céljából. Azonban még a jelölt de novo variánsok kimutatásakor is elégtelen információ áll rendelkezésre a gyakori és ritka variánsokról, ami kizárja az azonosított de novo variáns patogenitására és a betegségben betöltött szerepére vonatkozó egyértelmű következtetés levonását (Acuna-Hidalgo és mtsai., 2016). Ez a korlátozás azzal magyarázható, hogy a de novo variánsok általában heterozigóták, és lehetnek rendkívül ritkák vagy gyakoriak. A nagyon ritka de novo variánsok esetében a variáns patogenitását nehéz lehet bizonyítani, mivel nincs több olyan beteg, akinek azonos fenotípusa és de novo variánsa van. A gyakori de novo variánsok esetében előfordulhat, hogy a variáns patogenitásának megnyilvánulásait meghatározó tényezők nem ismertek, különösen akkor, ha az általános populáció néhány egyede rendelkezik a variánssal, de nem szenved a genetikai betegségben. A de novo variánsok arányától függetlenül azonban mindkét variánstípus skálázható a relatív fitnesz és a természetes szelekció alapján.
Az adaptivitás számos tényezőtől függ, ezért annak megítéléséhez, hogy egy DNM patogén vagy adaptív-e, és annak megértéséhez, hogy miért fordul elő egy adott gyakorisággal a populációban, a variánst megfelelő körülmények között kell vizsgálni. Ezek közé tartozik a környezet, a szülők életkora, a genomiális kontextus, az epigenetika és más tényezők, mivel ezek mind befolyásolják az átlagos relatív fitnesz értékét, amely monoton növekszik, miközben a szelekció erőssége csökken (Peck és Waxman, 2018).
A vizsgálat fő célja az volt, hogy tisztázzuk az előforduló DNM-ek előfordulási arányát, és meghatározzuk, hogyan oszlanak meg ezek a mutációk az általános litván populáció exomjában. Azt is megvizsgáltuk, hogy e mutációk gyakoriságát befolyásolja-e a szekvenciák összetétele vagy szerkezeti paraméterei, amelyekben előfordultak, valamint egyéb olyan tényezők, amelyek befolyásolhatják az e DNM-ek kialakulásának hátterében álló mechanizmusokat. Végül arra törekedtünk, hogy megállapítsuk, hogy a DNM-ek a funkcionális régiókra gyakorolt intenzív természetes szelekciós nyomás miatt alakultak-e ki. Bár a DNM-ek eloszlása és intenzitása már számos tanulmány tárgya volt, a litván populációban korábban még nem vizsgálták őket.
Anyagok és módszerek
A tanulmányban a LITGEN projekt (LITGEN, 2011) keretében kapott litván populációból származó mintákat elemeztünk. Az adatsor 49 trióból állt, összesen 144 különböző egyeddel. A genomi DNS-t vénás vérből vontuk ki fenol-kloroform extrakciós módszerrel vagy a paramágneses részecskék módszerén alapuló TECAN Freedom EVO® (Tecan Schweiz AG, Svájc) automatizált DNS extrakciós platformmal. Az exomokat SOLiD 5500 szekvenáló rendszerrel szekvenálták (75 bp olvasatok). A szekvenálási adatokat a Lifescope szoftverrel dolgoztuk fel és készítettük elő. Az exomokat a 19. számú humán referencia genom alapján térképeztük le. A szekvenálás átlagos olvasási mélysége 38,5 volt. Az anya, az apa és a gyermek Lifescope által generált BAM-formátumú fájljait SAMtools szoftverrel kombináltuk minden egyes trióhoz.
A de novo mutációkat két szoftverrel azonosítottuk: VarScan (Koboldt et al., 2012) és VarSeqTM. Egy potenciális variánst akkor tekintettünk DNM-nek, ha az utódokban azonosítottuk, de a szülők egyikében sem volt jelen ugyanazon a pozícióban. Összességében a VarScan és a VarSeqTM segítségével 1 752, illetve 4 756 DNM-et detektáltak. A hamis pozitív de novo hívások elvetése érdekében, amikor nem lehetett tudni, hogy a trió összes egyedét helyesen azonosították-e, konzervatív szűrőket alkalmaztunk a detektált DNM minőségi paraméterekre az alábbiak szerint: (1) az egyed genotípusminősége ≥50; (2) az egyes helyeken található leolvasások száma >20. Az SnpSift szoftvert használtuk e szűrők alkalmazására a VarScan által generált adatokon. A VarSeqTM szoftver által generált adatokat a Trio Workflow szegmensben ugyanezen szűrési paraméterek kiválasztásával szűrtük. Továbbá a fennmaradó szomatikus (csak a szekvenált vérsejtek egy töredékében jelen lévő), alacsony allélegyensúlyú vagy szekvenálási artefaktumokkal rendelkező variánsok elvetése érdekében a DNM-eket úgy szűrtük, hogy a trió esetében a leolvasásoknak az alternatív alléllal rendelkező egyénekben megfigyelt hányadára (az allélegyensúlyra) vonatkozó küszöbértéket állítottunk be (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Ezenkívül az összes lehetséges azonosított és szűrt de novo egynukleotid variánst kézzel felülvizsgáltuk az Integrative Genomics Viewer (Robinson et al., 2011) segítségével. Az azonosított DNM-ek nagy száma miatt a variánsok Sanger-szekvenálással történő validálásához 51 de novo egynukleotid variánst választottunk ki véletlenszerűen. A Sanger-szekvenálást ABI PRISM 3130xl genetikai analizátorral végeztük. A VarScan (N = 95) és a VarSeqTM (N = 84) által azonosított összes szűrt és kézzel felülvizsgált DNM-et ANNOVAR (Butkiewicz és Bush, 2016; Wang et al., 2010) segítségével annotáltuk. A fehérjék kölcsönhatásainak elemzéséhez a STRING szoftvert (Szklarczyk et al., 2017) használtuk. Az exomtérképezéshez hasonlóan az annotációkat a hg19 referencia humán genom felhasználásával végeztük el.
A valószínűséget, hogy egy hívó pozíció DNM volt a trióban, minden trió esetében egymástól függetlenül számoltuk ki. Egy korábbi hivatkozásban leírtak szerint (Besenbacher et al., 2015), a pozíciónkénti de novo arányt generációnként (PPPG) a következőképpen számoltuk ki:
ahol f a triók száma és N a hívható helyek száma, amelyek potenciálisan de novo helyként azonosíthatók minden trió esetében külön-külön, a szekvenálási mélységtől függetlenül. Ez a szám triótól függően változik. ni az i trióhoz azonosított DNM-ek száma. A Pji (de novos ingle nucleotide) valószínűségét, hogy a j és az i családba tartozó j egyetlen nukleotidhely mutálódik, a következőképpen számoltuk ki:
A Pji (de novo indel)valószínűségét a j nevű indel-hely és az i család mutációjára a következőképpen számoltuk:
ahol C, M és F az utódot, az anyát, illetve az apát jelenti, a Hetero, HomR és HomA pedig a heterozigóta, a referenciára homozigóta, illetve az alternatív allélra homozigóta allélt. A Pij (de novo) valószínűséget a szekvenálási lefedettség függvényében számoltuk ki. Az aránybecslések konfidenciaintervallumait a binomiális arányokhoz hasonlóan számoltuk ki. A DNM arány becsléséhez és a további számításokhoz az R csomagot (3.4.3 verzió) használtuk (R Core Team, 2013).
Azért, hogy teszteljük azt a hipotézist, hogy a genom különböző régióiban a DNM-ráta eltérései magyarázhatók-e magának a genomi régiónak a belső jellemzőivel és a szülő korával, lineáris regressziós elemzést végeztünk, amelyhez az egyes DNM-ek “másodlagos” annotációját az ENCODE (ENCODE Project Consortium, 2012) és LITGEN (LITGEN, 2011) projektek adatai alapján végeztük el. Először egy korábbi tanulmánynak (Besenbacher et al., 2015) megfelelően (ENCODE Project Consortium, 2012) lymphoblastoid sejtvonalakat (LCL és GM12878) választottunk az azonosított DNM-ek genomikai tájára vonatkozó rekordok összegyűjtéséhez. Adatokat gyűjtöttünk a:
(1) expressziós arányokra (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) vonatkozóan különböző szövetekben. A DNM-ekkel rendelkező régiók expressziója szerint specifikus és nem specifikus expresszióval rendelkező pozíciókat osztottunk;
(2) DNase1 hiperszenzitivitási helyek (DHS) mérése. A DHS-státuszt 0-val jelöltük, ha a DHS-csúcson kívül volt, és 1-gyel, ha azon belül;
(3) a CpG-szigetek kontextusának mérése. Ha a DNM a CpG-szigeteken belül volt, a pozíció státuszát 1; ha kívül – 0;
(4) három hisztonjel (H3K27ac, H3K4me1 és H3K4me3) az ENCODE projektből. Ha a DNM a hisztonnal jelölt pozícióban volt, akkor 1, ha nem – 0;
(5) GERPP++ konzervációs értékeket gyűjtöttünk ANNOVAR annotációs eszközzel. A konzervációs értékek alapján a DNM-ekkel rendelkező pozíciókat konzervatív (GERP++ score >12) és nem konzervatív (GERP++ score <12) pozíciókba soroltuk (Davydov et al., 2010; ENCODE Project Consortium, 2012). A LITGEN projekt kérdőíves nyilvántartásai alapján gyűjtöttük a szülők életkorára vonatkozó adatokat. A paraméterek összegyűjtése után minden egyes trió esetében kiszámították az egyes paraméterekkel rendelkező pozíciók számát. Ezután korrelációs elemzést, majd lineáris regressziós modellezést végeztünk a DNM-ráta és a paraméterek között.
Eredmények
A DNM-elemzést követően két trió (4. és 21. sz.) esetében kivételesen magas számú DNM-et azonosítottunk: 113 és 123 (VarScan és VarSeqTM segítségével), illetve 16 (VarScan). Ezek az eredmények arra késztettek bennünket, hogy megvizsgáljuk a biológiai apaságot, amit a trió esetében elutasítottunk. 4. számú trió esetében elutasítottuk, a 4. számú trió esetében pedig megerősítettük. 21. Így az adatok a trió No. adatokat kizártuk a vizsgálatból. A 48 trióból álló végső csoportban 34 trióban 95 DNM-et azonosítottunk a VarScan szoftverrel, 31 trióban pedig 84 DNM-et a VarSeqTM szoftverrel (1. ábra). A VarScan és a VarSeqTM szoftverrel 18, illetve 15 trióban nem észleltek DNM-et. A mindkét szoftver által azonosított DNM-ek közül csak a DNM-ek 5,37%-a egyezett (három DNM a MEIS2, PGK1 és MT1B génekben). Minden személyre átlagosan 1,9 (VarScan szoftver) és 1,7 (VarSeqTM) DNM jutott.
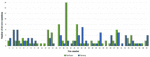
1. ÁBRA. A VarScan (kék) és a VarSeqTM (zöld) szoftverrel azonosított de novo egynukleotid variánsok összehasonlítása.
A VarScan szoftverrel azonosított 95 DNM elemzése azt mutatta, hogy 20 DNM exonikus volt, köztük két stop-gain DNM, hét szinonim DNM és 11 nem szinonim DNM. A VarSeqTM által azonosított 80 új mutáció exonikus volt, köztük 1 stop-gain DNM és 78 nem szinonim DNM (2. ábra). A VarScan által azonosított DNM-ek többsége az 1., 2., 4. és 5. kromoszómán volt, míg a VarSeqTM túlnyomórészt a 2., 6., 7. és 11. kromoszómán azonosított DNM-eket. Az azonosított DNM-ek száma nem korrelált a gének sűrűségével a kromoszómákon (R = 0,09, p-érték = 0,65 a VarScan esetében és R = 6,73, p-érték = 0,51 a VarSeqTM esetében) vagy a kromoszóma méretével (3. ábra). Mindkét szoftverprogram szerint az átmenetek és a transzverziók aránya nagyon hasonló volt: 1,44, illetve 1,47 (4. ábra). Az átmenetek szerkezetében azonban különbségeket azonosítottunk. Konkrétan, a VarScan által azonosított DNM-ek között több G/T és A/C átmenet volt, míg a VarSeqTM által azonosított DNM-ek között több A/T és G/C átmenet volt.
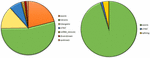
2. ÁBRA. A VarScan (balra) és a VarSeqTM (jobbra) által generált de novo mutációk (DNM) összetétele.
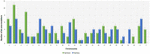
3. ábra. A de novo variánsok számának megoszlása kromoszómánként a VarScan és a VarSeqTM által generált adatok szerint. A zöld sávok a VarScan szoftver által azonosított DNM-eket, a kék sávok a VarSeqTM által azonosított DNM-eket jelölik. A hibasávok az egyes kromoszómák átlagos DNM-értékeinek standard hibáját jelölik.
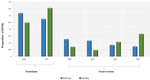
4. ÁBRA. Az átmenetek alapjául szolgáló molekuláris események gyakrabban fordulnak elő, mint a transzverziókhoz vezető események, ami azt eredményezi, hogy az egész exomban ∼1,5-szer nagyobb az átmenetek aránya a transzverziókhoz képest. A VarScan (zöld) és VarSeqTM (kék) szoftverek által azonosított átmenet és transzverzió események. A hibasávok az átlagos DNM-ek standard hibáját jelölik.
A de novo egynukleotid mutációk számított aránya a VarSeqTM szerint 2,4 × 10-8 PPPG (95%-os konfidenciaintervallum : 1,96 × 10-8-2,99 × 10-8), a VarSeqTM pedig 2.74 × 10-8 per nukleotid per generáció (95% CI: 2,24 × 10-8-3,35 × 10-8) a VarScan szerint.
A VarScan algoritmus három de novo indelt azonosított három trióban a 6. és 11. kromoszómában. A de novo indelek számított aránya a genomban 1,77 × 10-8 (95% CI: 6,03 × 10-9-5,2 × 10-8) PPPG volt. Figyelemre méltó, hogy minden de novo indel “reverzibilis” volt, azaz a szülők új variánsok voltak a genomban, gyermekeik pedig a referencia genom alapján a szekvenálási mélység 37,5 átlagértékével és 50 genotípusminőséggel rendelkező de novo variánsokkal rendelkeztek. Ezt a három DNM-et azonban nem választották ki a Sanger-szekvenálási módszerrel történő validálásra, így a de novo indelek túlbecslésének valószínűsége ennek ellenére megmarad. A de novo indelek C/T és A/G voltak az egyes nukleotidok kontextusában.
A lineáris regressziós modellezés kimutatta, hogy a DNSse 1 hiperszenzitivitási helyek, a CpG-szigetek kontextusa, a GERPP++ konzervációs értékek és az expressziós szintek ∼68-93%-ban magyarázták a DNM-ek arányát (1. táblázat). Sem az epigenetikai markerek, sem az apai életkor nem korrelált szignifikánsan a DNM-aránnyal. A modelleket csak a VarScanból nyert adatokból állítottuk fel, mivel nem volt korreláció a VarSeqTM adatai és magának a genomi régiónak a belső jellemzői között.

TABLE 1. TÁBLA. A DNAaseI hiperszenzitivitási helyek, a CpG-szigetek kontextusa, a GERPP++ konzervációs értékek és az expressziós szint hatásának lineáris regressziója a DNM-ek arányára.
A DNM-ek funkcionális előrejelzése
Azért, hogy felmérjük, mely miszenzmutációk voltak károsak és változtatták meg az érintett fehérje funkcióját típusonként, elemeztük a DNM-ek által kiváltott károsodások előrejelzett kategorikus pontszámát. A következő 10 értéket vettük figyelembe: polifen HDIV és HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, Fathmm-MKL kódolás és GERP++. Az előrejelzett pontszámok alapján a VarScan által azonosított négy olyan DNM-et választottunk ki, amelyek hat vagy több káros vagy valószínűleg káros előrejelzést tartalmaznak. Ezek a stop-gain DNM-ek a MEIS2 és ULK4 génekben, míg a nem szinonim DNM-ek az MT1B és PGK1 génekben voltak. Az e gének által kódolt fehérjék fontosak a neuronális növekedés, az endocitózis és a nehézfémek negatív hatásaival szembeni védelem szempontjából. Ezek a fehérjék részt vesznek a daganatos érgátló angiosztatin felszabadításában és különböző jelátviteli útvonalakban. Az e gének által kódolt fehérjék között nem volt kapcsolat (5. ábra).
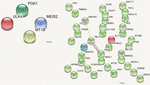
5. ábra. Fehérje-fehérje kölcsönhatások (Szklarczyk et al., 2017) a DNM-et hordozó génekben. Balra a VarScan által azonosított DNM-ek a fehérjéket kódoló génekben, jobbra a VarSeqTM által azonosított DNM-ek. A színes vonalak a fehérjék közötti kapcsolatot jelzik.
A VarSeqTM által azonosított de novo mutációkat részletesebben elemeztük, ha azokat a predikciós eszközök legalább fele károsnak vagy valószínűleg károsnak jósolta. 35 pontmutáció volt (lásd ??) olyan génekben, amelyek olyan fehérjéket kódolnak, amelyek fontosak a kromatin átalakításában, a citoszkeleton szabályozásában, a sejtnövekedésben és életképességben, a citoplazmatikus jelátviteli útvonalakban és a szaglás érzékelését kiváltó neuronális válaszok beindításában.
A DNM-érintett gének által kódolt fehérjék közül csak a CLPTM1, a ZNF547 és a DMXL1 kapcsolódott valamilyen módon (5. ábra).
Diszkusszió
A jelen tanulmányban átfogó elemzést végeztünk a DNM-ek eloszlásáról az exom különböző régióiban a litván populációban. A SOLiD 5500 szekvenálási technológia segítségével 34 trióban 95 DNM-et, illetve 31 trióban 84 DNM-et detektáltunk a VarScan, illetve a VarSeqTM algoritmus segítségével. Először is szeretnénk megjegyezni, hogy azért választottuk a VarScan-t a DNM-ek hívására, mert (Warden et al., 2014) szerint ez az algoritmus olyan variánslistát eredményez, amely nagyfokú (>97%) egyezést mutat a GATK UnifiedGenotyper és HaplotypeCaller által hívott, jó minőségű variánsokkal. A VarSeqTM szoftvert azért választottuk, mert széles körben használt eszköz a variánselemzéshez mind a kutatásokban, mind a klinikai elemzésben. Annak ellenére, hogy mindkét algoritmust úgy tervezték, hogy olyan DNM-eket keressen az utódok exomjában, amelyek egyik szülőben sem voltak jelen, a két szoftver DNM-elemzésében az egyezés a két szoftver között mindössze 5,37% volt. A VarScan algoritmusnak nagyobb volt az érzékenysége (5,42%) a DNM-ek szűrés előtti kimutatására, mint a VarSeqTM algoritmusnak (1,77%), így azt gyanítottuk, hogy egyik eszköz sem volt sikeres a mutációk hívásában a magas érzékenység miatt, amelyhez mindig alacsony specificitás társult. Ezért azt javasoljuk, hogy a különböző eszközök kimenetének kombinálásával jelentős javulást lehet elérni az eredményekben (Sandmann et al., 2017).
A generált adatok alapján a becsült egynukleotid DNM ráta 2,4 × 10-8 és 2,74 × 10-8 között volt, a de novo indeleké pedig 1,77 × 10-8 PPPG volt, az alkalmazott algoritmustól függően. Az általunk számított DNM-ráta magasabb volt, mint a korábbi tanulmányokban (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015) közöltek, amelyekben 1,2 × 10-8 és 1,5 × 10-8 PPPG között változott. A mi vizsgálatunkban mért magasabb DNM arány azért volt indokolt, mert vizsgálatunk exom adatokon alapult. Ráadásul az exomokban lényegesen magasabb (30%-kal) a mutációs ráta, mint a teljes genomokban, mivel a teljes genom bázispár-összetétele eltér az exomokétól. Nevezetesen, az exomok átlagos GC-tartalma körülbelül 50%, míg a teljes genomé körülbelül 40% (Neale et al., 2012). A metilált CpG-k az emberben a citozin bázisok spontán deaminációja miatt erősen mutálódó szekvenciákat képviselnek (Neale et al., 2012). Összehasonlító genomikai vizsgálatok szerint a CpG-gazdag régiókban megnövekedett mutációs arányok feltehetően az emlősök sugárzása körül alakultak ki (Francioli et al., 2015). A fajok divergenciája során a CpG-gazdag exonikus régiók a nem kódoló DNS-hez képest megnövekedett mutációs rátán mentek keresztül, és nem kódoló régiókká alakultak. Ezért aztán a CpG-tartalom hatása idővel csökken, az átlagos mutációs ráta csökken, amíg el nem éri a környező nem kódoló DNS-ben jelen lévő szintet (Subramanian és Kumar, 2003). Míg azonban a genom semlegesen fejlődő régióiban lévő szekvenciáknak elegendő idejük volt arra, hogy a dinukleotid-kontextusok tekintetében egyensúlyba kerüljenek, a tisztító szelekció fenntartotta a funkcionális régiókban a hipermutábilis CpG-ket (Subramanian és Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Ezért, mivel magasabb DNM-arányt találtunk, mint amiről más tanulmányok beszámoltak, azt feltételeztük, hogy ez legalább részben a helyi szekvencia-kontextusnak és/vagy az exomra gyakorolt esetleges természetes szelekciós nyomásnak tudható be. Ennek megfelelően lineáris regressziós modellt alkalmaztunk, és azt találtuk, hogy a DNAse 1 hiperszenzitivitás, a CpG-szigetek kontextusa, a GERPP++ konzervációs értékek és az expressziós szint a DNM-ráta ∼68-93%-át magyarázta. Ezek az eredmények azt jelezték, hogy az exomban a DNM-ek a DNS-szekvenciák konzerváltságától függetlenül alakultak ki. A DNM-ráta azonban magasabb volt azokban a génekben, amelyek termékei nem specifikusak voltak, valamint a transzkripcionálisan aktív promóterszerű régiókban.
Más tanulmányok eredményeivel ellentétben (Wong és mtsai., 2016; Sandmann és mtsai., 2017) azt találtuk, hogy az apai életkor nem korrelált a DNM-ráta mértékével. Ezek az eredmények azzal magyarázhatók, hogy az adathalmaz hasonló szülői életkorú triókból állt, és hogy a teljes genomnak csak egy kis részét (∼1,5%) elemeztük. Ezen paraméterek alapján minden egyes személynek átlagosan csak 1,9 (VarScan) vagy 1,7 (VarSeqTM) DNM-je volt, míg a teljes genomban 40-82 (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al, 2016), míg a kódoló szekvenciában a de novo indelek száma hasonló volt a (Front Line Genomics, 2017) által azonosítottakhoz.
Az annotációk kiterjedt funkcionális elemzésének eredményei azt mutatták, hogy az összes azonosított DNM közül 4 (VarScan) és 35 (VarSeqTM) variáns valószínűleg patogén DNM volt. A patogén DNM-ek számában mutatkozó különbség azzal magyarázható, hogy a DNM-ek azonosítására használt algoritmustól függően a DNM-ek aránya a kódoló szekvenciákban jelentősen különbözött. Például a VarScan szoftverrel azonosított DNM-ek 21,05%-a exonikus volt, míg a VarSeqTM szoftverrel azonosítottak 95,24%-a exonikus. Ezek a patogén DNM-ek a kromatin modellezéséhez, a citoszkeleton szabályozásához, a sejtnövekedés és a vitalitás modulálásához, a citoplazmatikus jelátviteli útvonalak működéséhez és a neuronális válasz beindításához elengedhetetlen fehérjéket kódoló génekben voltak. Annak ellenére, hogy ezeket a DNM-eket patogénnek tekintették, a felmérésben részt vevő összes egyén genetikailag “egészségesnek” azonosította magát. Ez az eredmény tehát azt jelezte, hogy a DNM-ek feltételezett patogenitása ellenére a genomok, amelyekben a DNM-ek elhelyezkedtek, nyilvánvalóan tolerálták ezeket a változásokat, így a betegség manifesztációi gyakran nem voltak kifejezettek. Szamecz és munkatársai (2014) szerint minél gyakrabban fordulnak elő DNM-ek konzervált genetikai pozíciókban, annál erősebb a természetes szelekció hatása a genetikai változásokra a genomvédelem kompenzációs mechanizmusain keresztül. A variánsok káros hatásait négyféleképpen lehet mérsékelni. Egyes gének tolerálják a fehérjék csonka változatait, mert funkcionális hatásukat elfedik a hiányos expresszió, a kompenzáló változatok vagy a csonkolás alacsony funkcionális jelentősége (Bartha és mtsai., 2015). Ezzel szemben a nem szinonim DNM-ekkel járó génváltozásokat az egész genomban hasznos mutációk felhalmozódásának mechanizmusa kompenzálja (Szamecz és mtsai., 2014). Ez arra utal, hogy ezekben az esetekben a patogén mutációk nem elég károsak ahhoz, hogy csökkentsék az átlagos fittséget, és ezért a természetes szelekció által alakítva sok generáción keresztül hosszabb ideig fennmaradnak.
Összefoglalva, a DNM-ek eloszlásának, valamint genetikai és epigenetikai kontextusuknak az elemzése betekintést nyújtott a litván genom genetikai variációjába. Ezen eredmények alapján a genetikai betegségekben szenvedő betegcsoportokon végzett további vizsgálatok megkönnyíthetik, hogy bizonyos patogén DNM-eket meg tudjunk különböztetni a tolerált háttér-DNM-ektől, és megbízható okozó DNM-eket tudjunk azonosítani. E tanulmány fő korlátja azonban az volt, hogy nem vizsgáltuk a nem kódoló és szabályozó génrégiók variációját. Ez az információ hozzájárulhatna a DNM-ek kialakulásának lehetséges mechanizmusainak tisztázásához, amelyek még mindig nem eléggé tisztázottak.
Accession Codes
A szekvenciaadatokat az Európai Nukleotid Archívumban (ENA) helyeztük el, a PRJEB25864 (ERP107829) hozzáférés alatt.
Etikai nyilatkozat
Ezt a vizsgálatot a Vilnius Regional Ethics Committee for Biomedical Research engedélyezési ajánlásainak megfelelően végeztük. A protokollt a Vilnius Regionális Biomedikai Kutatási Etikai Bizottság jóváhagyta. Minden alany a Helsinki Nyilatkozatnak megfelelően írásbeli beleegyezését adta.
Author Contributions
LP végezte az adatelemzést és készítette a kéziratot. AJ kiszámította a de novo mutációk arányát. A trió exomok szekvenálását LA és IK végezte. VK volt a vezető kutató.
Finanszírozás
Ezt a tanulmányt az Európai Szociális Alap támogatta a Global Grant intézkedés keretében. LITGEN projekt sz. VP1-3.1-ŠMM-07-K-01-013.
Érdekütközésre vonatkozó nyilatkozat
A szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségként értelmezhetők.
Kiegészítő anyagok
A cikkhez tartozó kiegészítő anyagok online elérhetők a következő címen: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). Új betekintés a de novo mutációk keletkezésébe és szerepébe az egészségben és a betegségben. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). A heterozigóta fehérjecsonkoló variánsok jellemzői a humán genomban. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Új variáció és de novo mutációs ráták populációszerte de novo összeállított dán triókban. Nat Commun. 6:5969. doi: 10.1038/ncomms696969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). CpG-sziget-klaszterek és proepigenetikus szelekció a HOX és más transzkripciós faktorok fehérjekódoló exonjaiban található CpG-k számára. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). A genomi variáció in silico funkcionális annotációja. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Az emberi spontán mutáció eredete, mintái és következményei. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). A GERP++ segítségével a humán genom nagy hányadának azonosítása, amely szelektív korlátozás alatt áll. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Az emberi genom DNS-elemeinek integrált enciklopédiája. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., and Keightley, P. D. (2007). Az új mutációk fitneszhatásainak eloszlása. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). A de novo mutációk genomszintű mintázatai és tulajdonságai az emberben. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics Magazine 14. szám – ASHG. London: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Genetikai hatások a génexpresszióra emberi szövetekben. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: szomatikus mutációk és kópiaszám-változások felfedezése rákban exom szekvenálással. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). A de novo mutációk aránya és az apa életkorának jelentősége a betegség kockázatában. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Finomszintű rekombinációs sebességbeli különbségek nemek, populációk és egyedek között. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ‘t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). A transzkriptom és genom szekvenálás funkcionális variációt tár fel az emberben. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Elérhető a következő címen: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Az exonikus de novo mutációk mintázata és aránya autizmus spektrumzavarokban. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Mi az adaptáció és hogyan kell mérni? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). A Language and Environment for Statistical Computing. Bécs: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integratív genomikai néző. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Variánshívó eszközök értékelése nem illesztett újgenerációs szekvenálási adatokhoz. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., and Sunyaev, S. (2008). A hipermutálható nem szinonim helyek erősebb negatív szelekció alatt állnak. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). A főemlősök genomjában a neutrális szubsztitúciók gyorsabban fordulnak elő az exonokban, mint a nem kódoló DNS-ben. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovács, K., Fekete, G., Farkas, Z., et al. (2014). A kompenzációs evolúció genomikai tájképe Be. A kompenzációs evolúció genomikai tájképe. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). A STRING adatbázis 2017-ben: minőség-ellenőrzött fehérje-fehérje asszociációs hálózatok, széles körben hozzáférhetővé téve. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: genetikai variánsok funkcionális annotálása újgenerációs szekvenálási adatokból. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Két népszerű variánshívó csomag részletes összehasonlítása exom- és célzott exonvizsgálatokhoz. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Új megfigyelések az anyai életkor csíravonalbeli de novo mutációkra gyakorolt hatásáról. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar