A vesekőbetegség előfordulása és prevalenciája továbbra is növekszik a lakosság körében. Az akár 10%-os életprevalenciával a nephrolithiasis (NL) és a nephrocalcinosis (NC) ezért jelentős egészségügyi terhet jelent, különösen a nyugati világban (1). Az NL és NC jelentős morbiditással jár, és a kiújulás, az ismétlődő sebészeti/endoszkópos beavatkozások és a kísérő gyulladás miatt krónikus vesebetegséggé alakul. Leegyszerűsítve, a vesekőképződés a vizeletben a kristályosodást gátló (pl. citrát, magnézium, uromodulin és pirofoszfát) és elősegítő (pl. oxalát, kalcium, foszfát, urát és cisztin) anyagok egyensúlyhiányából ered, ami meghaladja a szupertelítettséget, következetes aggregációval, nukleációval és a kő növekedésével a Randall-plakkban (1. ábra). Ez az egyensúlyhiány a promóterek vagy inhibitorok megváltozott enterális és/vagy renális kezelése miatt alakulhat ki, például az oxalát enterális malszekréciója vagy a kalcium renális malreabszorpciója miatt (1. ábra).

1. ábra. A vizeletben lévő kristályosodást gátló és elősegítő anyagok egyensúlyhiánya, ami vesekő kialakulásához vezet. A húgyúti inhibitorok és promotorok koncentrációját mind a bélrendszeri, mind a vese transzporterek befolyásolják és szabályozzák (GI-vese tengely). Ezek a transzporterek és cserélők, mint például az SLC26A1, felelősek a szekrécióért és a felszívódásért. Az oxalát tekintetében az enterális hiperabszorpció, de malszekréció és/vagy a renális hiperszekréció, de malabszorpció a vizelet oxalát szintjének a szupertelítettséget meghaladó szintjéhez vezet, és ezáltal elősegíti a kristályosodást és a konzekvens kőképződést .
A NL mögöttes etiológiája feltehetően multifaktoriális, környezeti, jelentős étrendi, hormonális és genetikai komponenssel. Ikervizsgálatokban a vesekő öröklődését 56%-ra becsülték (3), és a hypercalciurás kőbetegek akár kétharmadának van NL-ben szenvedő rokona (4). Bár a kalciumtartalmú vesekövek több mint 80%-át teszik ki, az ilyen kövek genetikai alapja továbbra is nagyrészt ismeretlen (5). A CLDN14, TRPV5, SLC34A1, ALPL, CASR és UMOD variánsaitól eltekintve a genom-széles körű asszociációs vizsgálatok még nem mutattak ki jelentős genetikai tényezőket (6-8). Rizikóallélokat azonosítottak azonban olyan géneken belül, amelyekről szintén kiderült, hogy a betegséget mendeli alapon továbbítják, mint például a CASR, az SLC34A1 és az SLC2A9 (9, 10). A mai napig több mint 30 olyan egyedi gént azonosítottak, amelyeknek Online Mendelian Inheritance in Man-defined fenotípusa mutáció esetén szerepet játszhat az NL/NC kialakulásában (1. táblázat).

Táblázat 1. táblázat. Az NL/NC monogénes formáit okozó ismert gének.
A monogénes formák öröklődési módja az autoszomális domináns, autoszomális-recesszív és X-kromoszómás öröklődés. Érdekes módon számos ilyen gén esetében mind recesszív, mind domináns öröklődési módról beszámoltak: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 és SLC4A1. Míg a legtöbb szindrómás és súlyos veleszületett rendellenesség recesszív öröklődési mintát mutat (Bartter, Lowe, Dent, FHHNC és distalis renális tubuláris acidózis szenzorineurális süketséggel), az enyhébb állapotok inkább domináns gének mutációihoz kapcsolódnak. A kódolt fehérjék többsége veseoldat-transzporter (pl. SLC34A1, SLC34A3 és SLC9A3R1), de kloridcsatornák (CLCN5), tight-junction fehérjék (pl. CLDN16/CLDN19) és metabolizáló enzimek (pl. AGXT, APRT és CYP24A1) defektusát is találták NL/NC-s betegekben. Ezért a mögöttes hiba többnyire magában a vese tubuláris rendszerében található, és ezért tubulopátiának tulajdonítható. Ezzel szemben eleve extrarenális állapotok, mint a primer hyperoxaluria (PH), ahol a májenzimek (AGXT, GRHPR és HOGA1) diszfunkciója oxalát-felhalmozódást okoz másodlagos veseérintettséggel, elképzelhető okai az NL/NC-nek. Bár mindegyik betegségfenotípusról úgy gondolják, hogy viszonylag ritka entitást képvisel, az egygénes okok széles genetikai heterogenitásuk révén jelentős számú beteget jelenthetnek (42). A genetikai heterogenitás mellett létezik allélvariáció is, ahol a csonka variánsok inkább funkcióvesztést eredményeznek, a missense variánsok (hipomorfok) pedig inkább finom defektusokat okozhatnak, amelyek klinikailag áttekinthetőek, különösen a felnőtt kőképzőkben. Egy másik, nemrégiben felismert jelenség a géndózishatásokról szól a fent említett vesekőgének közül több génben. Az SLC34A3 esetében például, amely a proximális tubulus egyik fő foszfáttranszporterét (NaPiIIc) kódolja, kimutatták, hogy a heterozigóta egyének már nem tekinthetők pusztán egészséges hordozóknak, mivel a vad típusú egyéneknél lényegesen gyakrabban mutatnak veseelmeszesedést és/vagy csontkinövést; de még mindig kisebb mértékben, mint a biallelikus (homozigóta és összetett heterozigóta) egyének (43). Hasonló megfigyelésekről számoltak be a CYP24A1 mutációival rendelkező családok esetében (44). A monogén rendellenességek hozzájárulását a vesekőbetegség általános gyakoriságához korábban nem vizsgálták átfogóan. Különösen hiányoznak a genetikai bizonyítékok, amelyek az okozó gének sokaságának széles körű szűrésén alapulnak nagy betegcsoportokban. Az átfogó genetikai vizsgálatok a múltban túl költségesek és nem voltak hatékonyak. A legtöbb NL/NC-ben szenvedő egyén esetében ezért nem volt elérhető az okozó genetikai hibára vonatkozó mutációelemzés, annak ellenére, hogy az NL/NC molekuláris okának ismerete fontos következményekkel járhat a prognózis, a profilaxis és/vagy a kezelés szempontjából. A klinikai megfigyelési vizsgálatokból csak durva becslések születtek: egy hatalmas adatgyűjtés alapján a kőösszetétel elemzéséből arra a következtetésre jutottak, hogy a monogén okok aránya gyermekeknél nem haladja meg a 9,6%-ot, felnőtteknél pedig az 1,6%-ot (45). Az elmúlt évtizedben azonban ez a helyzet a nagy áteresztőképességű szekvenálási technikák megjelenésével változni kezdett.
NL/NC-s betegek nagy áteresztőképességű mutációelemzése
A vesekőbetegségben szenvedő betegek vizsgálatára az ismert betegséggének patogén mutációinak jelenlétére vonatkozóan mikrofluidikus multiplex-PCR-en és konzekvens NextGen szekvenáláson (Fluidigm™/NGS) alapuló génpanelt hoztunk létre (46, 47).
Egy “pilot-tanulmányban” 268 genetikailag rendezetlen egyént toboroztunk konzekvensen tipikus veseköves klinikákról; 102 gyermek- és 166 felnőtt próbaidős személyt. Ennek eredményeként 30 elemzett génből 14-ben 50 károsító variánst azonosítottunk, ami az esetek 15%-ában molekuláris diagnózishoz vezetett. A gyermekkori alcsoportban 21%-ban mutattunk ki okozó mutációt, míg a felnőttek körében 11%-ban volt jelen delétező variáns (2A ábra) (48). Az SLC7A9 cystinuria-gén mutációit a felnőtt kohorszban találtuk a leggyakrabban (2B ábra). Két követéses vizsgálat is meg tudta erősíteni ezeket az eredményeket. Először is, egy 143 NL/NC-betegből álló, kizárólag gyermekkori kohorszban az esetek 17%-át 14 különböző gén mutációjával magyarázták (49). Másodszor, egy 51 családból álló kohorszban, ahol az NL/NC 25 éves kor előtt manifesztálódott, a célzott WES segítségével csaknem 30%-ban sikerült genetikai okot kimutatni (50). Nem meglepő módon a recesszív mutációkat gyakrabban találták újszülötteknél és veleszületett betegség esetén, míg a domináns állapotok általában az élet későbbi szakaszában manifesztálódtak. Ezek az adatok azt jelzik, hogy a genetikai eredetű vesekőbetegség aluldiagnosztizált állapot, annak ellenére, hogy a molekuláris diagnózis potenciálisan befolyásolja a prognózist, a profilaxist és/vagy a kezelést. Említésre méltó korlátozás azonban a lehetséges szelekciós torzítás, amely abból adódik, hogy mindhárom említett vizsgálatban veseköves szakrendelőkből toborozták a résztvevőket.

2. ábra. A 30 ismert monogénes nephrolithiasis (NL)/nephrocalcinosis (NC) gén mutációjának elemzése 268 NL/NC-s betegnél. (A) A monogén okok aránya a gyermek és felnőtt alkohortban. (B) A monogén okok száma a gének között (piros a felnőtteket, narancssárga a gyermekbetegeket jelöli). Megjegyzendő, hogy az SLC7A9-et találták a leggyakrabban mutálódottnak, különösen a fiatal felnőtteknél.
Identification of Novel Human Disease Genes by Candidate-Gene Approach
A nagy áteresztőképességű mutációelemzést különböző jelölt gének patogén variánsainak szűrésére is használják. A közelmúlt egyik legérdekesebb eredménye az SLC26A1 humán mutációinak felfedezése volt (32). Amióta Dawson és munkatársai 2010-ben először leírták a Ca-oxalát (CaOx) vesekő-képződést és NC-t Slc26a1 (Sat1)-knockout egerekben, az SLC26A1 jóhiszeműen NL-jelölt génnek számít (51). Az SLC26A1 egy anioncserélőt kódol, amely a proximális vesetubulusok, az ileum és a jejunum basolaterális membránján expresszálódik. Következésképpen a jelölt gén megközelítést alkalmazva patogén variánsokat azonosítottak olyan emberekben, akiknél a CaOx-NL korai kezdetben jelentkezett, nevezetesen két nem rokon személynél, akiknél biallelikus miszenzváltozatokat találtak (32). Funkcionálisan az azonosított variánsok patogenitását in vitro bizonyították, ami intracelluláris félrevezetéshez és károsodott transzportaktivitáshoz vezetett (32). A hibás SLC26A1 tehát a CaOx-NL új okát képezi, és figyelembe kell venni, amikor egyének vizsgálatát végezzük a visszatérő CaOx-kő képződés okainak feltárásakor.
Új klinikai betegregiszter az örökletes vesekőbetegséghez
A nyugati országokban növekvő prevalenciára vonatkozó legtöbb epidemiológiai adat amerikai adatbázisokból származik. Bár sürgősen szükség lenne rá, központosított európai adatbázisok jelenleg nem állnak rendelkezésre. Mivel az örökletes vesekőbetegség prevalenciájáról szóló, fent említett genetikai vizsgálatokat mind Európában, mind az USA-ban specializált központokból származó kis kohorszokkal végezték, az európai általános helyzetre való lefordítás nem érvényes. Míg az Egyesült Államokban a Ritka Vesekő Konzorcium olyan platformot alkot, amely integrálja és koordinálja az olyan ritka betegségekkel kapcsolatos nyilvántartási, alaptudományi és klinikai kutatási tevékenységeket, mint a cisztinúria, a PH, az APRT-hiány, a Dent- és a Lowe-kór, addig sem Európában, sem Németországban nem valósult meg hasonló adatgyűjtés az örökletes vesekőbetegségben szenvedő betegekről. A már létező európai PH-regiszterrel, az OxalEurope-pal (Prof. Bernd Hoppe, Bonni Egyetem) együttműködve, valamint a Deutsche Forschungsgemeinschaft és az Else Kröner-Fresenius Stiftung finanszírozásával a közelmúltban létrehoztuk a Lipcsei Egyetemen az “örökletes vesekőbetegség klinikai betegregiszterét”. A regisztert országos szinten a Német Felnőttnefrológiai Társaság (DGfN) és a Gyermeknefrológiai Társaság (GPN) támogatja. Továbbá a német klinikai vizsgálati regiszterbe is be van jegyezve (DRKS-ID: DRKS00012891). A vizsgálat toborzásának alapvető részeként az ismert és új vesekőgének nagy áteresztőképességű mutációelemzését kutatási alapon kínálják fel olyan betegek számára, akiknek nincs megalapozott molekuláris diagnózisa, de a klinikai kép alapján genetikai hajlamra utal: pl. korai életkor (<40 év), pozitív családi anamnézis, indikatív fenotípusok, mint NC, cisztinúria vagy RTA, és súlyosan visszatérő NL (>3×) (2. táblázat). Míg a már megállapított genetikai diagnózissal rendelkező betegeket általában bevonják, a másodlagos NL/NC okokkal, például malignitással, szarkoidózissal és primer hyperparathyreoidizmussal járó eseteket nem veszik be a genetikai elemzésbe. A betegek aktív felvételéhez egy klinikai központnak általában a helyi intézményi felülvizsgálati bizottság jóváhagyására van szüksége; ehhez a folyamathoz segítséget és támogatást nyújtunk a megfelelő sablonok biztosításával. Az etikai jóváhagyást követően a beleegyező nyilatkozat és a klinikai adatlapok (pl. klinikai kérdőív) letölthetők a regiszterünk honlapjáról (http://www.mks-registry.net). Az alapos klinikai fenotipizálás biztosítása érdekében olyan lényeges betegadatokat fogunk kérni, mint többek között az etnikai hovatartozás, a vérrokonság, a családi anamnézis, a betegség kezdetének kora, a kiújulás (minden feltételezett új veseköves eseményként definiálva), a napi folyadékbevitel, a sebészeti beavatkozások és a vesén kívüli érintettség. A dokumentumokat a beteg a beiratkozó orvos segítségével töltheti ki. Ezenkívül biokémiai szérumparamétereket, beleértve a kreatinin, eGFR, PTH, D-vitamin, elektrolitok, húgysav és vizeletvizsgálat (pH, kalcium, foszfát, magnézium, húgysav, citrát, oxalát és cisztin, lehetőleg 24 órás vizeletből, ha nem spot vizeletből), valamint a kőösszetétel elemzésének adatait kérik a beiratkozáskor. Figyelembe véve, hogy a 24 órás vizeletgyűjtést és a kőösszetétel-elemzést nem minden intézményben végzik rutinszerűen, ezeket a paramétereket a rendelkezésre állástól függően vesszük fel. Kívánatos, de nem kötelező a felvett betegek 2 éves klinikai utánkövetése. A regisztrációt követően a toborzó klinikai központok személyre szabott bejelentkezési lehetőséget kapnak a betegek adatainak a nyilvántartási honlapunkon (http://www.mks-registry.net) keresztül történő megadásához. Alternatívaként felajánljuk az adatok elektronikus úton történő bevitelét, ha azokat papír alapon küldik el nekünk. A bevitt adatokat egy biztonságos szerveren tároljuk, és a részt vevő klinikai központok hozzáférhetnek saját betegadataik megtekintéséhez. A következő weboldalakon további információk találhatók:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
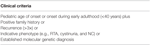
Táblázat 2. táblázat. A mutációelemzés felvételi kritériumai a klinikai betegregiszterben.
Összefoglalva, a vesekőbetegség egyre gyakoribb állapot, amely klinikailag heterogén és kevéssé ismert, különösen a genetikai mozgatórugói. Amint azt egy sor közelmúltbeli tanulmány jelezte, a monogén állapotok prevalenciáját nagy valószínűséggel alábecsülik. Az örökletes vesekőbetegségre vonatkozó központi betegnyilvántartás bevezetésével hozzájárulunk ahhoz, hogy legalább részben felszámoljuk a vesekőbetegség genetikájával kapcsolatos hatalmas ismerethiányt. Ebben az összefüggésben a klinikai regiszterek több okból is értékes források: először is, a fenotípus-genotípus összefüggések jobb meghatározása kulcsfontosságú lesz a betegek pontosabb rétegzéséhez a jövőbeli klinikai kutatások során. Másodszor, új betegséggének azonosítása új betegségmechanizmusokkal csökkenti az ismeretlen NL/NC etiológia hiányát; és harmadszor, az új molekuláris célpontok megfejtése segít a súlyosan érintett családokban a kiújulást megelőző gyógyszerek kifejlesztésében.
Etikai nyilatkozat
Ezt a vizsgálatot az “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” ajánlásainak megfelelően, valamennyi alany írásbeli beleegyezésével végeztük. Minden alany a Helsinki Nyilatkozatnak megfelelően írásos beleegyezését adta. A protokollt az “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” hagyta jóvá.”
Author Contributions
JH fogalmazta meg és írta a kéziratot. BH, AS, LM és RS szerkesztették a kéziratot, és felépítették a regiszter infrastruktúráját, amelyet bemutatnak az olvasónak.
Enyilatkozat az összeférhetetlenségről
A szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségként értelmezhetők.
Finanszírozás
A kutatást a DFG (HA 6908/2-1) és az EKFS (2016_A52) által JH-nak nyújtott projekttámogatás finanszírozza. Ezt a munkát továbbá a németországi Szövetségi Oktatási és Kutatási Minisztérium (BMBF) támogatta, FKZ: 01EO1501 JH-nak.
1. Scales CD Jr., Smith AC, Hanley JM, Saigal CS. A vesekövek prevalenciája az Egyesült Államokban. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. A hypercalciurás nephrolithiasis genetikája: vesekőbetegség. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Előrelépés a kalciumtartalmú nefrolithiasis genetikájának megértésében. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576.
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Az abszorpciós hiperkalciuria fenotípushoz és az alacsony gerincvelői csontsűrűséghez társuló báziscseréket tartalmazó gén azonosítása és jellemzése. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. A peroxiszomális L-alanin:glioxilát aminotranszferáz mitokondriumokba történő hibás célzása primer hiperoxalúriás betegekben egy kriptikus mitokondriális célszekvencia pontmutáció általi aktiválásától függ. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Humán adenin-foszforibozil-transzferáz. Allélmutációk azonosítása nukleotid szinten az enzim teljes hiányának okaként. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutations in ATP6N1B, encoding a kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Egy belga családban a karbonátanhidráz II deficiencia szindrómát egy pontmutáció okozza egy invariáns hisztidin maradékon (107 His→Tyr): a normál humán CA II gén teljes szerkezete. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A calcium-sensing receptor mutációi miatt kialakuló családi hipokalcémia szindróma hypercalciuriával. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. A common molecular basis for three inherited kidney stone diseases. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, egy vese tight junction protein, amely szükséges a paracelluláris Mg2+ reszorpcióhoz. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Autoszomális recesszív FAM20A mutációk által okozott nefrokalcinózis (zománcveseszindróma). Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. A hidroxipiruvát-reduktázt (GRHPR) kódoló gén mutálódik a II. típusú primer hiperoxalúriában szenvedő betegekben. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. A HNF4A R76W mutáció a β-sejt fenotípus mellett atipikus domináns Fanconi szindrómát okoz. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. 17 független mutáció azonosítása, amelyek a humán hipoxantin-guanin-foszforibozil-transzferáz (HPRT) hiányáért felelősek. Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnion, átmeneti antenatális Bartter-szindróma és MAGED2 mutációk. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. A Lowe oculocerebrorenális szindróma szorosan kapcsolódó flanking markerei, alkalmazással a hordozói állapot felmérésére. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. A Bartter-szindróma, hipokalaemiás alkalózis hiperkalciuriával, a Na-K-2Cl kotranszporter NKCC2 mutációi okozzák. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutations in SLC26A1 cause nephrolithiasis. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinuria okozta mutációk az rBAT-ban, a cisztin transzportjában részt vevő génben. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Az rBAT egyik alegységét (bo,+AT) kódoló SLC7A9 mutációi által okozott nem I-es típusú cisztinúria. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutációk és a renális parathormon érzékenysége. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Sugallt bizonyíték egy fogékonysági génre a D-vitamin-receptor lókusz közelében az idiopátiás kalciumkőképződésben. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Két mutáció azonosítása a humán xantin-dehidrogenáz génben, amely a klasszikus I. típusú xanthinuriáért felelős. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Hildebrandt F. Genetikus vesebetegségek. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Monoallelikus és biallelikus CYP24A1 mutációkkal rendelkező felnőttek klinikai és biokémiai fenotípusai: a géndózishatás bizonyítékai. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Récents acquis diagnostiques et thérapeutiques. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. 99 új mutáció azonosítása egy 1056 nephronophthisisszel összefüggő ciliopathiás betegből álló világszintű kohorszban. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Monogén okok prevalenciája nephrolithiasisban vagy nephrocalcinosisban szenvedő gyermekbetegeknél. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar