

Alecensa (alectinib) is een kinaseremmer bestemd voor de behandeling van patiënten met anaplastisch lymfoom kinase (ALK)-positieve niet-kleincellige longkanker (NSCLC).
Het geneesmiddel is ontwikkeld door Roche Group-lid Genentech in samenwerking met Chugai Pharmaceutical.
Alecensa ontving initiële goedkeuring in Japan in juli 2014, en van de Amerikaanse Food and Drug Administration (FDA) in december 2015.
Roche diende in september 2015 een aanvraag voor een vergunning voor het in de handel brengen in bij het Europees Geneesmiddelenbureau (EMA) voor de goedkeuring van Alecensa.
Het bedrijf ontving in februari 2017 een voorwaardelijke vergunning voor het in de handel brengen van Alecensa als monotherapie van de Europese Commissie (EC) voor de behandeling van volwassen patiënten met ALK-positief gevorderd NSCLC.
Deze patiënten werden eerder behandeld met crizotinib, een NSCLC-geneesmiddel ontwikkeld door Pfizer.
ALK-positief gevorderd NSCLC
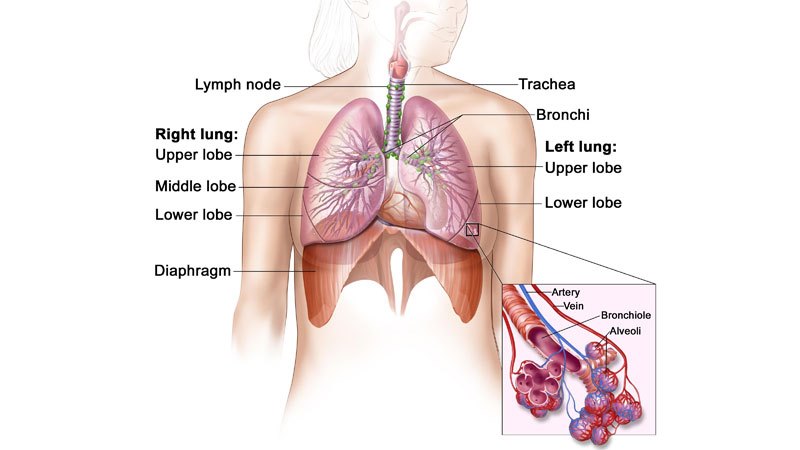
Niet-kleincellig longcarcinoom is het meest voorkomende type kanker, en maakt meer dan 85% van de longkankers uit. NSCLC leidt tot de dood van ongeveer 1,59 miljoen mensen per jaar wereldwijd.
De ziekte ontstaat wanneer cellen van de long abnormaal worden en zich oncontroleerbaar beginnen te ontwikkelen. De diagnose wordt meestal in een vergevorderd stadium gesteld, en staat erom bekend dat de ziekte in een vroeg stadium moeilijk op te sporen of te diagnosticeren is.
Symptomen die met longkanker worden geassocieerd, zijn hoesten, kortademigheid, vermoeidheid, gebrek aan eetlust, en gewichtsverlies.
ALK-positieve NSCLC komt voor bij ongeveer 5% van de patiënten die lijden aan gevorderd NSCLC, met naar schatting 75.000 mensen per jaar die wereldwijd worden gediagnosticeerd.
Alecensa’s werkingsmechanisme
Alecensa bevat een tyrosinekinaseremmer, die ALK-fosforylering en ALK-gemedieerde activering van de downstream signaleringseiwitten voorkomt die in NSCLC-tumoren zijn geïdentificeerd.
Het geneesmiddel is momenteel beschikbaar in de vorm van capsules van 150 mg voor orale toediening.
Klinische proeven met Alecensa
De voorwaardelijke goedkeuring van de EG voor het in de handel brengen van Alecensa was gebaseerd op twee klinische fase I / II-studies, genaamd NP28673 en NP28761.
De NP28673 klinische studie was een fase I / II wereldwijde single-arm, open-label multi-center studie, die de veiligheid en werkzaamheid van Alecensa evalueerde bij 138 ALK-positieve NSCLC-patiënten van wie de ziekte was gevorderd op crizotinib.
Resultaten toonden aan dat patiënten die werden behandeld met Alecensa een overall respons rate (ORR) van 50 vertoonden.8% in een beoordeling door een onafhankelijke beoordelingscommissie, die werd gemeten aan de hand van de Response Evaluation Criteria In Solid Tumors (RECIST)-criteria.
Een beoordeling door een onderzoeker toonde aan dat tumoren afnamen bij 51,4% van de mensen die Alecensa kregen.
De patiënten bleven reageren gedurende een mediane periode van 15,2 maanden, terwijl de mediane progressievrije overleving (PFS) bij mensen die Alecensa kregen 8,9 maanden bedroeg.
Resultaten toonden ook aan dat het veiligheidsprofiel van Alecensa vergelijkbaar was met het profiel dat in eerdere studies werd waargenomen.
De bijwerkingen die werden gemeld bij ≥2% van de patiënten die tijdens de studie met het geneesmiddel werden behandeld, omvatten dyspneu, anemie, vermoeidheid, INR verhoogd, longembolie en hyperbilirubinemie.
NP28761 was een fase I / II klinische studie uitgevoerd in Noord-Amerika. Het was een single-arm, open-label multicenteronderzoek dat de veiligheid en werkzaamheid van Alecensa evalueerde bij 87 ALK-positieve NSCLC-patiënten, van wie de ziekte ook progressie had vertoond tijdens de behandeling met crizotinib.
De resultaten van de studie toonden aan dat patiënten die werden behandeld met Alecensa een ORR van 52,2% vertoonden in een beoordeling door een onafhankelijke beoordelingscommissie, gemeten aan de hand van de RECIST-criteria.
Een beoordeling door de onderzoeker toonde aan dat tumoren in 52,9% afnamen na behandeling met het geneesmiddel.
De proefpersonen bleven reageren gedurende een mediaan van 14,9 maanden, en de mediane PFS bij mensen die Alecensa kregen was acht maanden.
Resultaten toonden aan dat het veiligheidsprofiel van Alecensa vergelijkbaar was met het profiel dat in eerdere studies werd waargenomen.
De meest voorkomende graad 3 of hoger bijwerkingen die tijdens de klinische studie werden gemeld, waren onder meer toename van spierenzymen, toegenomen leverenzymen, kortademigheid, verhoogde triglycerideniveaus, lage fosfaat- en kaliumniveaus, en tijd verlengd voor gedeeltelijke bloedverdikking.