Alexei Drummond, Andrew Rambaut, Remco Bouckaert, and Walter Xie
1 Introduction
Deze tutorial introduceert de BEAST software voor Bayesiaanse evolutionaire analyse aan de hand van een eenvoudige tutorial. De tutorial omvat co-estimatie van een gen fylogenie en bijbehorende divergentie tijden in de aanwezigheid van kalibratie-informatie van fossiele bewijzen.
U dient te beschikken over de volgende software:
- BEAST – dit pakket bevat het BEAST-programma, BEAUti, TreeAnnotator en andere hulpprogramma’s. Deze handleiding is geschreven voor BEAST v2.2.x, dat ondersteuning biedt voor meerdere partities. Het kan worden gedownload van http://www.beast2.org/.
- Tracer – dit programma wordt gebruikt om de uitvoer van BEAST (en andere Bayesiaanse MCMC-programma’s) te onderzoeken. Het vat de verdelingen van continue parameters grafisch en kwantitatief samen en verschaft diagnostische informatie. Op het moment van schrijven is de huidige versie v1.6. Deze kan worden gedownload van
http://tree.bio.ed.ac.uk/software/. - FigTree – dit is een toepassing voor het weergeven en afdrukken van moleculaire fylogenieën, in het bijzonder die welke zijn verkregen met behulp van BEAST. Op het moment van schrijven is de huidige versie v1.4.2. Het kan worden gedownload van http://tree.bio.ed.ac.uk/software/.
Deze tutorial leidt u door de analyse van een alignment van sequenties van twaalf primatensoorten (zie figuur 1). Het doel is een schatting te maken van de fylogenie, de evolutiesnelheid van elke lineage en de ouderdom van de niet-gekalibreerde stralingsdivergenties.
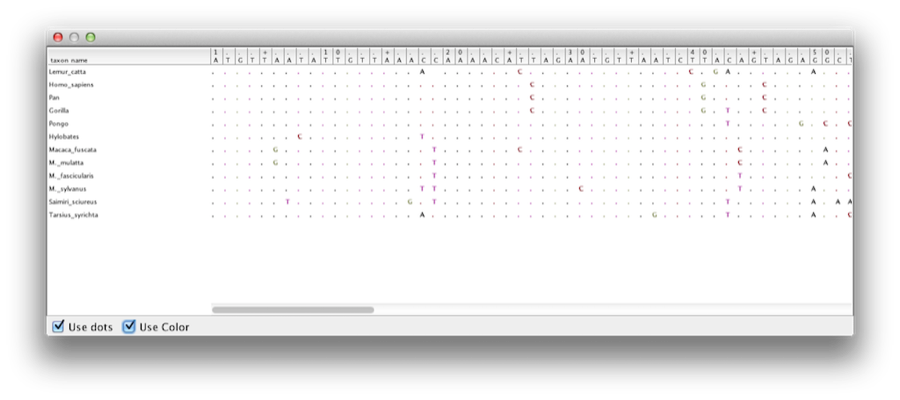
Figuur 1: Een deel van de uitlijning voor primaten.
De eerste stap is het omzetten van een NEXUS-bestand met een DATA- of CHARACTERS-blok in een BEAST XML-invoerbestand. Dit wordt gedaan met het programma BEAUti (dat staat voor Bayesian Evolutionary Analysis Utility). Dit is een gebruikersvriendelijk programma voor het instellen van het evolutionaire model en opties voor de MCMC-analyse. De tweede stap is het daadwerkelijk uitvoeren van BEAST met behulp van het door BEAUTi gegenereerde invoerbestand, dat de gegevens, het model en de analyse-instellingen bevat. De laatste stap is het onderzoeken van de uitvoer van BEAST om problemen vast te stellen en de resultaten samen te vatten.
2 BEAUti
Het programma BEAUti is een gebruikersvriendelijk programma voor het instellen van de modelparameters voor BEAST. Start BEAUti door te dubbelklikken op het pictogram ervan. Als BEAUti eenmaal draait, ziet het er ongeveer hetzelfde uit, ongeacht het computersysteem waarop het draait. Voor deze handleiding wordt de Mac OS X versie gebruikt in de figuren, maar de Linux en Windows versies zullen dezelfde layout en functionaliteit hebben.
2.1 Het laden van het NEXUS bestand
Om een uitlijning in NEXUS formaat te laden, selecteert u eenvoudig de Import Alignment… optie uit het File menu, of sleept u het bestand naar het midden van Partitions panel.
Het voorbeeldbestand met de naam primate-mtDNA.nex is beschikbaar in de map examples/nexus/ voor Mac en Linux en examples/nexus/ voor Windows in de map waarin BEAST is geïnstalleerd. Dit bestand bevat een uitlijning van sequenties van 12 primatensoorten.
Een Add Partition venster (Figuur 2) verschijnt als het betreffende pakket is geïnstalleerd. Als u “zuiver” BEAST 2 gebruikt, kunt u naar de volgende paragraaf gaan. Selecteer anders Add Alignment en klik op OK om verder te gaan.
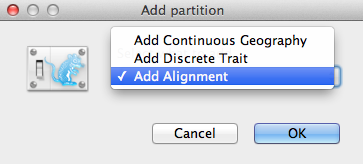
Figuur 2: Venster Add Partition (Partitie toevoegen) (verschijnt alleen als verwante pakketten zijn geïnstalleerd).
Als de codering in de partities overlappingen vertoont, verschijnt het venster met waarschuwingsberichten (Figuur 3). Lees en klik op OK om verder te gaan.
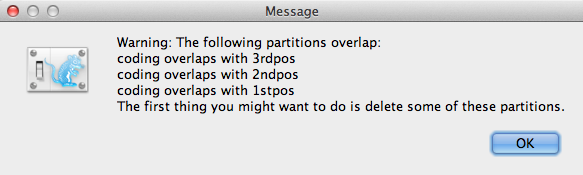
Figuur 3: Venster met waarschuwingsberichten (verschijnt alleen als er overlappingen in de partities zijn wat betreft codering).
Eenmaal geladen, worden er vijf tekenpartities weergegeven in het hoofdpaneel (Figuur 4). De uitlijning is verdeeld in een eiwitcoderend deel en een niet-coderend deel, en het coderende deel is verdeeld in codonposities 1, 2 en 3. U moet de ‘coding’ partitie verwijderen voordat u verder gaat met de volgende stap, omdat deze verwijst naar dezelfde nucleotiden als partities ‘1stpos’, ‘2ndpos’ en ‘3rdpos’. Om de ‘coding’ partitie te verwijderen selecteert u de rij en klikt u op de ‘-‘ knop onderaan de tabel. U kunt de uitlijning bekijken door te dubbelklikken op de partitie.
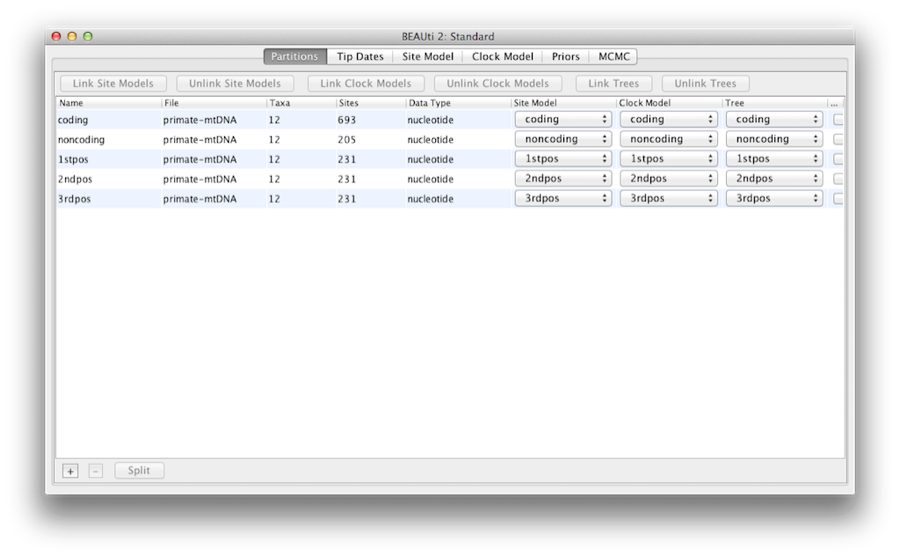
Figuur 4: Een screenshot van het gegevenstabblad in BEAUti. Deze en alle volgende screenshots zijn gemaakt op een Apple-computer met Mac OS X en zullen er op andere besturingssystemen iets anders uitzien.
Partitiemodellen koppelen/ontkoppelen
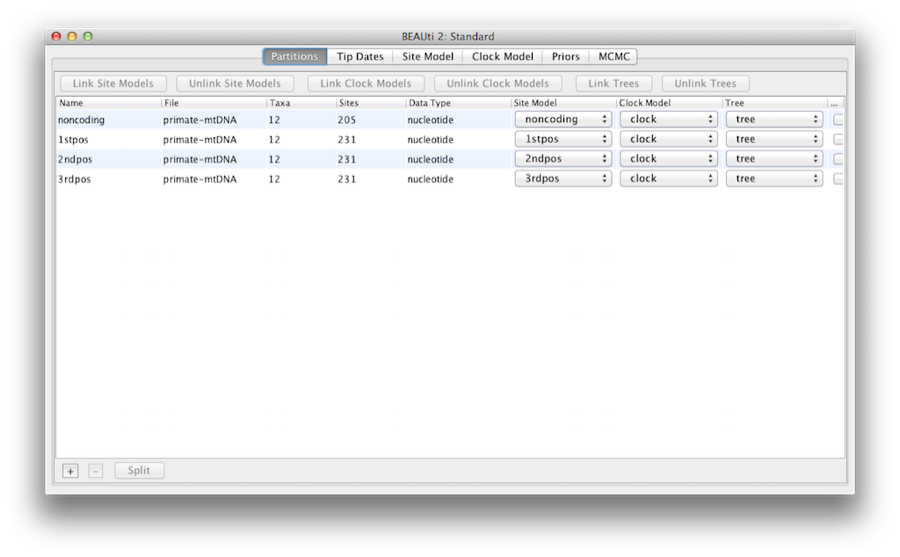
Figuur 5: Een screenshot van het tabblad Partities in BEAUti na het koppelen en hernoemen van het klokmodel en de boom.
Omdat de sequenties gekoppeld zijn (d.w.z. dat ze allemaal afkomstig zijn van het mitochondriale genoom, waarvan wordt aangenomen dat het bij vogels en zoogdieren geen recombinatie ondergaat), hebben ze dezelfde voorouders, zodat de partities dezelfde tijdboom in het model zouden moeten hebben. Omwille van de eenvoud zullen we ook aannemen dat de partities dezelfde evolutiesnelheid hebben voor elke tak, en dus hetzelfde “klokmodel”. We zullen onze modellering van de snelheid heterogeniteit beperken tot heterogeniteit tussen sites binnen elke partitie, en ook toestaan dat de partities verschillende gemiddelde snelheden van evolutie hebben.
Dus, op dit punt moeten we het klokmodel en de boom koppelen. Selecteer in het Partitions-paneel alle vier de partities in de tabel (of geen enkele, standaard zijn alle partities betrokken) en klik op de knop Link Trees en vervolgens op de knop Link Clock Models (zie figuur 5). Klik dan op het eerste drop-downmenu in de kolom Klokmodel en hernoem het gedeelde klokmodel naar ‘clock’. Hernoem op dezelfde manier de gedeelde boom in ’tree’. Dit maakt de volgende opties en de gegenereerde logbestanden gemakkelijker leesbaar.
2.2 Instellen van het substitutiemodel
De volgende stap is het instellen van het substitutiemodel. Selecteer vervolgens het tabblad Site Models bovenaan het hoofdvenster (we slaan het tabblad Tip Dates over omdat alle taxa afkomstig zijn van contemporaine monsters). Dit zal de evolutionaire modelinstellingen voor BEAST onthullen. De beschikbare opties hangen af van de vraag of de gegevens nucleotiden, aminozuren, binaire gegevens of algemene gegevens zijn. De instellingen die worden weergegeven na het laden van de primaten-nucleotidenuitlijning zijn de standaardwaarden voor nucleotidengegevens, dus we moeten enkele wijzigingen aanbrengen.
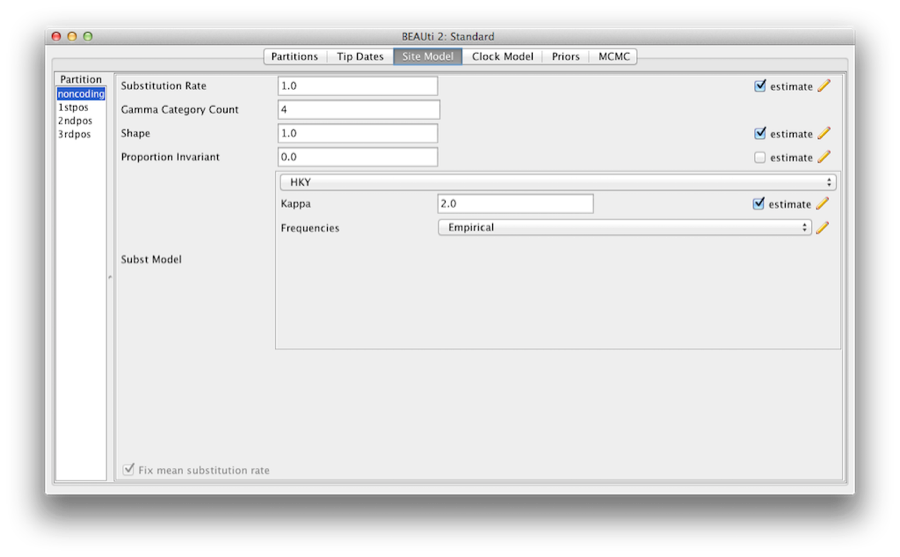
Figuur 6: Een screenshot van het tabblad Site Model in BEAUti.
De meeste modellen zullen u bekend voorkomen. Stel eerst de Gamma Categorie Count in op 4 en vink vervolgens het vakje “estimate” aan voor de Shape-parameter. Hierdoor kan de variatie in snelheid tussen de locaties in elke partitie worden gemodelleerd. Merk op dat 4 tot 6 categorieën goed genoeg is voor de meeste datasets, terwijl meer categorieën meer tijd kosten om te berekenen voor weinig extra voordeel. We laten het item Proportion Invariant op nul staan.
Selecteer vervolgens HKY uit het Subst Model drop-down menu. Idealiter zou een substitutiemodel moeten worden gekozen dat voor elke partitie het best bij de gegevens past, maar hier gebruiken we omwille van de eenvoud HKY voor alle partities. Kies voorts Empirisch in het vervolgkeuzemenu Frequenties. Hierdoor worden de frequenties vastgesteld op de in de gegevens waargenomen verhoudingen (voor elke partitie afzonderlijk, zodra we de site-modellen loskoppelen). Deze aanpak betekent dat we een goede fit met de gegevens kunnen krijgen zonder deze parameters expliciet te schatten. We doen dat hier alleen om de logbestanden wat korter en leesbaarder te maken in latere delen van de exercitie.
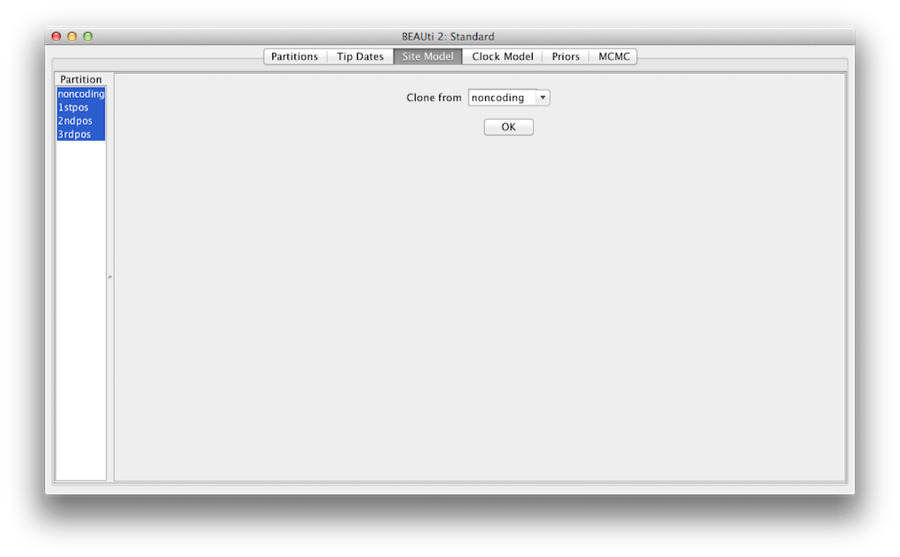
Figuur 7: kloonconfiguratie van één sitemodel naar andere.
Vink ten slotte het vakje ‘schatten’ aan voor de parameter Substitutiesnelheid en schakel het selectievakje Gemiddelde mutatiesnelheid vastzetten in. Hierdoor kunnen voor de afzonderlijke partities de relatieve snelheden worden geschat voor de niet-gekoppelde site-modellen (figuur 6).
Ten slotte houdt u de ‘shift’-toets ingedrukt om alle site-modellen aan de linkerkant te selecteren en klikt u op OK om de instelling van niet-coderend naar 1stpos, 2ndpos en 3rdpos te klonen (figuur 7). Ga door elk site model, zoals u kunt zien, hun configuraties zijn nu hetzelfde.
2.3 Het klokmodel instellen
De volgende stap is het selecteren van het Klokmodellen tabblad bovenaan het hoofdvenster. Hier selecteren we het moleculaire klokmodel. Voor deze oefening laten we de selectie op de standaardwaarde van een strikte moleculaire klok, omdat deze gegevens zeer klokachtig zijn, en geen snelheidsvariatie tussen takken nodig hebben om in het model te worden opgenomen.
Om te testen of het model op de klok lijkt, kun je (i) de analyse uitvoeren met een ontspannen klokmodel en nagaan hoeveel variatie tussen de snelheden de gegevens impliceren (zie variatiecoëfficiënt voor meer hierover), of (ii) een modelvergelijking uitvoeren tussen een strikte en ontspannen klok met behulp van pathsampling, of (iii) een willekeurig lokaal klokmodel gebruiken dat expliciet nagaat of elke tak in de boom zijn eigen aftakkingssnelheid nodig heeft.
2.4 Priors
Op het tabblad Priors kunnen priors worden gespecificeerd voor elke parameter in het model. De modelselecties die zijn gemaakt in de tabbladen Locatiemodel en Klokmodel, resulteren in de opname van verschillende parameters in het model, en deze worden weergegeven in het tabblad Priors (zie Figuur 8).
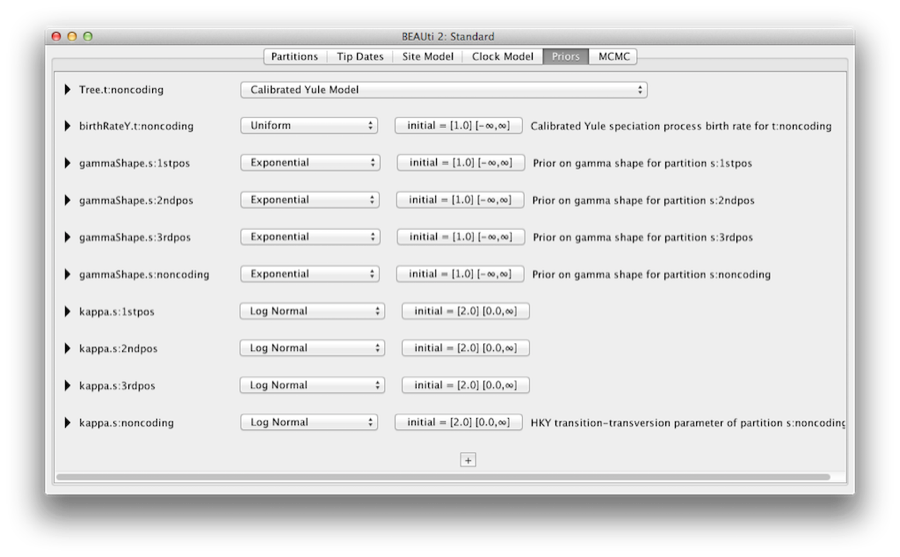
Figuur 8: Een screenshot van het tabblad Priors in BEAUti.
Hier geven we ook aan dat we het gekalibreerde Yule-model als boomprior willen gebruiken. Het Yule-model is een eenvoudig model van soortvorming dat over het algemeen beter geschikt is wanneer sequenties van verschillende soorten worden bekeken. Selecteer het Gekalibreerde Yule Model in het Boomvoorafgaande dropdown menu.
2.4.1 Definiëren van de ijkknoop
We moeten nu een prior distributie specificeren op de gekalibreerde knoop, gebaseerd op onze fossiele kennis. Dit staat bekend als het kalibreren van onze boom. Om een extra prior te definiëren, druk op de kleine + knop onder de lijst van priors. Als het niet zichtbaar is in uw view, scroll dan naar beneden in het paneel om de + knop te vinden. U zult een dialoogvenster zien waarmee u een subset van de taxa in de fylogenetische boom kunt definiëren. Als u eenmaal een taxaverzameling hebt gemaakt, kunt u later kalibratie-informatie toevoegen voor de meest recente commonancestor (MRCA).
Noem de taxa set door het invullen van het taxon set label. Noem het mens-chimpansee, omdat het de taxa voor Homo sapiens en Pan zal bevatten. In de lijst hieronder zie je de beschikbare taxa. Selecteer achtereenvolgens elk van de twee taxa en druk op de pijlknop > >. (Figuur 9).Klik OK en de nieuw gedefinieerde taxa set wordt toegevoegd aan de prior lijst.Omdat dit een gekalibreerde node is die gebruikt moet worden in combinatie met de Gekalibreerde Yule prior, moet monofylie worden afgedwongen, dus vink het vakje Monofyletisch aan. Hierdoor wordt de boomtopologie zodanig beperkt dat de groep mens- chimpansee gedurende de MCMC-analyse monofyletisch blijft.
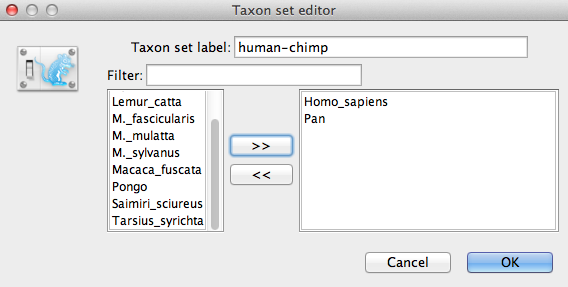
Figuur 9: Taxonset-editor in BEAUti.
Om de kalibratie-informatie te coderen, moeten we een verdeling opgeven voor de MRCA van mens-chimpansee.Selecteer de lognormale verdeling uit het vervolgkeuzemenu rechts van de nieuw toegevoegde mens-chimpansee.prior. Klik op het zwarte driehoekje en een grafiek van de kansdichtheidsfunctie verschijnt, samen met de parameters voor de lognormale verdeling. Wij gaan M=1.78 en S=0.085 instellen, hetgeen een verdeling oplevert met als middelpunt ongeveer 6 miljoen jaar en een standaardafwijking van ongeveer 0.5 miljoen jaar. Dit geeft een centrale 95% waarschijnlijkheidsrange die 5-7 Mya omvat. Dit komt ruwweg overeen met de huidige consensusschatting van de datum van de meest recente gemeenschappelijke voorouder van mensen en chimpansees (Figuur 10).
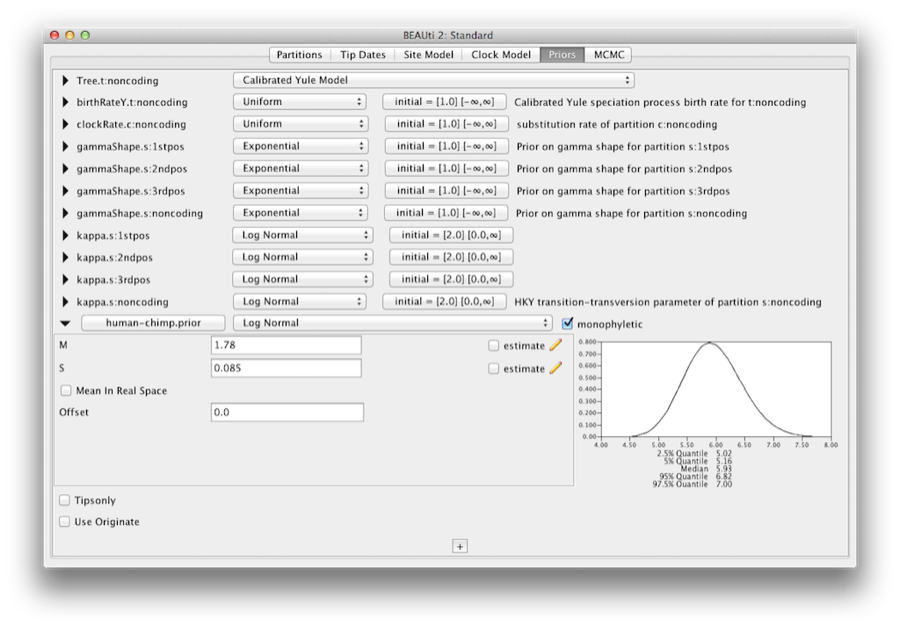
Figuur 10: Een screenshot van de opties voor de ijkingsprioriteit in het paneel Priors in BEAUti.
We moeten ons ervan overtuigen dat de in het priorpaneel getoonde priors werkelijk de prior-informatie weerspiegelen die we hebben over de parameters van het model. Tenslotte zullen we ook enkele diffuse “niet-informatieve” maar juiste priors specificeren voor de algemene moleculaire kloksnelheid (clockRate) en de speciatiesnelheid (birthRateY) van de Yule tree prior. Selecteer voor elk van hen Gamma uit het drop-down menu en gebruik de pijlknop om het beeld uit te breiden om de parameters van de Gamma prior te onthullen. Stel voor zowel de klokfrequentie als de Yule geboortefrequentie de parameter Alpha (vorm) in op 0.001 en de parameter Beta (schaal) op 1000 (Figuur 11).
Bestandaard heeft elk van de gamma-vormparameters een exponentiële prioriteitsverdeling met een gemiddelde van 1. Dit impliceert (zie Figuur 3.7) dat we een geringe variatie verwachten. Standaard hebben de kappa-parameters voor het HKY-model een lognormale(1,1.25) prioriteitsverdeling, die in grote lijnen overeenkomt met empirisch bewijs voor het bereik van realistische waarden voor overgangs-/overgangsbias. Deze standaard priors worden aangehouden omdat ze geschikt zijn voor deze specifieke analyse.
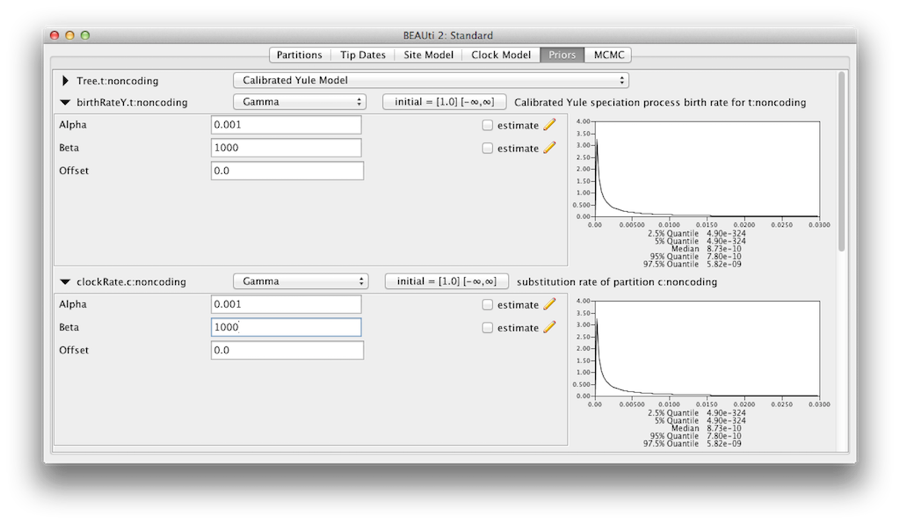
Figuur 11: Gamma prior.
2.5 De MCMC-opties instellen
Het volgende tabblad, MCMC, bevat meer algemene instellingen om de lengte van de MCMC-run en de bestandsnamen te regelen.
Eerst hebben we de ketenlengte. Dit is het aantal stappen dat de MCMC in de keten zal maken voordat hij eindigt. Hoe lang dit moet zijn hangt af van de grootte van de gegevensverzameling, de complexiteit van het model en de kwaliteit van het gewenste antwoord. De standaardwaarde van 10.000.000 is volledig willekeurig en moet worden aangepast aan de grootte van uw gegevensverzameling. Voor deze gegevensverzameling stellen we de kettinglengte in op 6.000.000, omdat dit op de meeste moderne computers redelijk snel zal gaan (een paar minuten).
Het veld Store Every bepaalt hoe vaak de toestand in een bestand wordt opgeslagen. Het periodiek opslaan van de status is handig voor situaties waarin de computeromgeving niet erg betrouwbaar is en een BEAST run kan worden onderbroken. Met een opgeslagen kopie van de recente toestand kunt u de keten hervatten in plaats van opnieuw te beginnen vanaf het begin, zodat u niet opnieuw door burn-in hoeft te gaan. Het veld Pre Burnin geeft het aantal monsters aan dat niet wordt gelogd aan het begin van de analyse. We laten de velden Store Every en Pre Burnin op hun standaardwaarden staan. Daaronder staan de details van de logbestanden. Elk bestand kan worden uitgebreid door op het zwarte driehoekje te klikken.
De volgende opties geven aan hoe vaak de parameterwaarden in de Markovketen op het scherm moeten worden getoond en in het logbestand moeten worden vastgelegd.De schermuitvoer is alleen bedoeld om de voortgang van het programma te volgen en kan op elke waarde worden ingesteld (als deze te klein wordt ingesteld, zal de hoeveelheid informatie die op het scherm wordt weergegeven het programma vertragen). Voor het logbestand moet de waarde worden ingesteld in verhouding tot de totale lengte van de keten. Te vaak bemonsteren leidt tot zeer grote bestanden met weinig extra voordeel voor de nauwkeurigheid van de analyse. Als te weinig wordt bemonsterd, wordt in het logbestand niet voldoende informatie over de verdeling van de parameters opgenomen. Je wilt waarschijnlijk niet meer dan 10.000 monsters opslaan, dus dit moet worden ingesteld op niet minder dan ketenlengte / 10.000.
Voor deze oefening stellen we de frequentie van het sporenlogbestand en het bomenlogbestand in op 1.000 en die van het schermlogbestand op 10.000.Geef ook Primates.log op als de bestandsnaam van het sporenlogbestand en Primates.trees als de bestandsnaam van het bomenlogbestand.Zorg ervoor dat de bestandsnaam van het schermlog leeg blijft, anders wordt het schermlog niet naar het scherm geschreven.
- Als je het Windows-besturingssysteem gebruikt, raden we je aan het achtervoegsel .txt toe te voegen aan beide bestanden (dus Primates.log.txt en Primates.trees.txt), zodat Windows deze herkent als tekstbestanden.
2.6 Het genereren van het BEAST XML-bestand
We zijn nu klaar om het BEAST XML-bestand te maken. Selecteer hiervoor de optie Opslaan in het menu Bestand. Controleer de standaardprincipes en sla het bestand op met een geschikte naam (we sluiten de bestandsnaam meestal af met .xml, bijvoorbeeld Primates.xml). We zijn nu klaar om het bestand door BEAST te halen.
3 BEAST uitvoeren
Figuur 12: Een screenshot van BEAST.
Draai nu BEAST en wanneer het om een invoerbestand vraagt, geeft u uw nieuw aangemaakte XML-bestand op als invoer. BEAST zal dan draaien totdat het klaar is met het rapporteren van informatie naar het scherm. De eigenlijke resultatenbestanden worden op de schijf opgeslagen op dezelfde plaats als uw invoerbestand. De uitvoer naar het scherm zal er ongeveer zo uitzien:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Merk op dat er in het begin nuttige informatie staat over de alignementen en welke boomwaarschijnlijkheden zijn gebruikt. Ook worden alle citaten die relevant zijn voor de analyse aan het begin van de run vermeld, die gemakkelijk kunnen worden gekopieerd naar manuscripten waarin over de analyse wordt gerapporteerd. Daarna volgt de rapportage van de keten, die enige real time feedback geeft over de voortgang van de keten.
Aan het eind wordt een operator-analyse afgedrukt, die alle in de analyse gebruikte operatoren opsomt, samen met hoe vaak de operator werd geprobeerd, geaccepteerd en afgewezen (zie respectievelijk de kolommen #total, #accept en #reject). Het aanvaardingspercentage is het percentage keren dat een exploitant wordt aanvaard wanneer hij wordt geselecteerd om een voorstel te doen. In het algemeen wijst een hoog aanvaardingspercentage, bijvoorbeeld meer dan 0,5, erop dat de voorstellen conservatief zijn en de parameterruimte niet efficiënt verkennen. Een laag aanvaardingspercentage daarentegen wijst erop dat de voorstellen te agressief zijn en bijna altijd resulteren in een toestand die wordt verworpen wegens zijn lage posterioriteit. Zowel te hoge als te lage aanvaardingspercentages resulteren in lage ESS-waarden. Een acceptatiegraad van 0,234 is het streefcijfer (gebaseerd op zeer beperkt bewijsmateriaal van ) voor veel (maar niet alle) operatoren die in BEAST zijn geïmplementeerd.
Sommige operatoren hebben een afstemmingsparameter, bijvoorbeeld de schaalfactor van de ascale parameter. Als het uiteindelijke acceptatiepercentage niet in de buurt van het doel ligt, zal BEAST een nieuwe waarde voor de afstemparameter voorstellen, die in de analyse van de operator wordt afgedrukt. In dit geval zijn alle acceptatiepercentages goed voor de operatoren die afstemparameters hebben. Operatoren zonder afstemmingsparameters zijn voor deze analyse onder andere de wide-exchange en Wilson-Balding operatoren. Beide operatoren proberen de topologie van de boom met grote stappen te veranderen, maar aangezien de gegevens overwegend een enkele topologie ondersteunen, worden deze radicale voorstellen bijna altijd verworpen.
4 Analyse van de resultaten
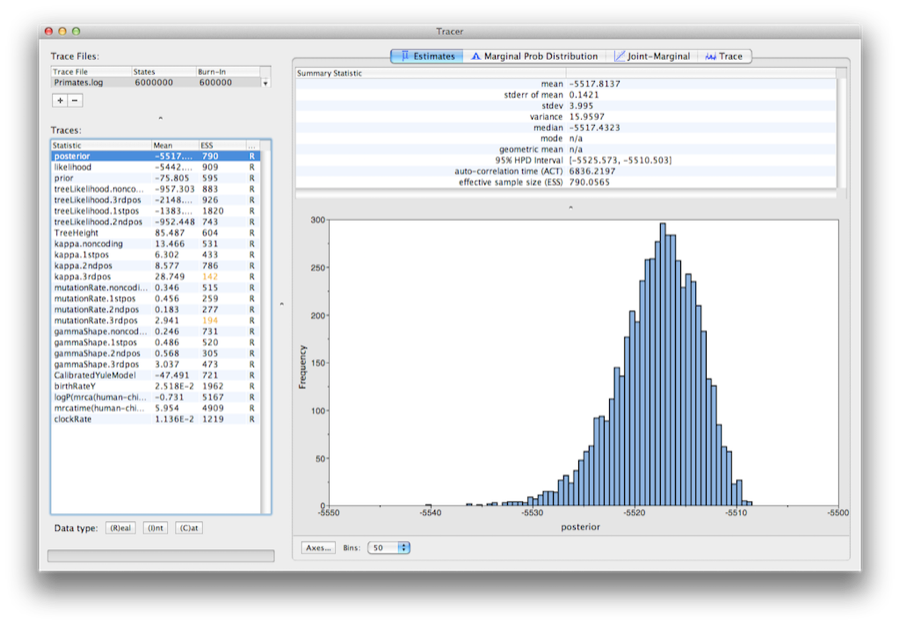
Figuur 13: Een screenshot van Tracer v1.6.
Run het programma met de naam Tracer om de uitvoer van BEAST te analyseren. Wanneer het hoofdvenster is geopend, kiest u in het menu Bestand de optie Sporenbestand importeren… en selecteert u het bestand dat BEAST heeft gemaakt met de naam Primaten.log (figuur 13).
Bedenk dat MCMC een stochastisch algoritme is, zodat de werkelijke getallen niet precies hetzelfde zullen zijn als die in de figuur.
Aan de linkerkant staat een lijst met de verschillende grootheden die BEAST naar een bestand heeft gelogd. Er zijn sporen voor de posterior (dit is de natuurlijke logaritme van het product van de boomwaarschijnlijkheid en de prioriteitsdichtheid), en de continue parameters. Het selecteren van een spoor aan de linkerkant brengt analyses voor dit spoor aan de rechterkant te voorschijn, afhankelijk van het tabblad dat is geselecteerd. Wanneer het voor het eerst wordt geopend, wordt het ‘posterior’ spoor geselecteerd en worden verschillende statistieken van dit spoor getoond onder het tabblad Estimates. Rechtsboven in het venster staat een tabel met berekende statistieken voor het geselecteerde spoor.
Selecteer de clockRate parameter in de linker lijst om te kijken naar de gemiddelde snelheid van evolutie (gemiddeld over de gehele boom en alle sites). Tracer zal een (marginaal posterior) histogram plotten voor de geselecteerde statistiek en zal ook samenvattende statistieken geven zoals het gemiddelde en de mediaan. De 95% HPD staat voor hoogste posterior density interval en vertegenwoordigt het meest compacte interval op de geselecteerde parameter dat 95% van de posterior waarschijnlijkheid bevat. Het kan losjes worden beschouwd als een Bayesiaans equivalent van een betrouwbaarheidsinterval. De TreeHeight parameter geeft de marginale posterior verdeling van de leeftijd van de wortel van de gehele boom.
Selecteer de TreeHeight parameter en Ctrl-klik dan op mrcatime(human-chimp) (Command-klik op Mac OS X). Dit toont een weergave van de leeftijd van de wortel en de kalibratie MRCA die we eerder in BEAUti hebben opgegeven. U kunt verifiëren dat de divergentie die we hebben gebruikt om de boom te kalibreren(mrcatime(human-chimp)) een posterior verdeling heeft die overeenkomt met de priorverdeling die we hebben opgegeven (afbeelding 14).
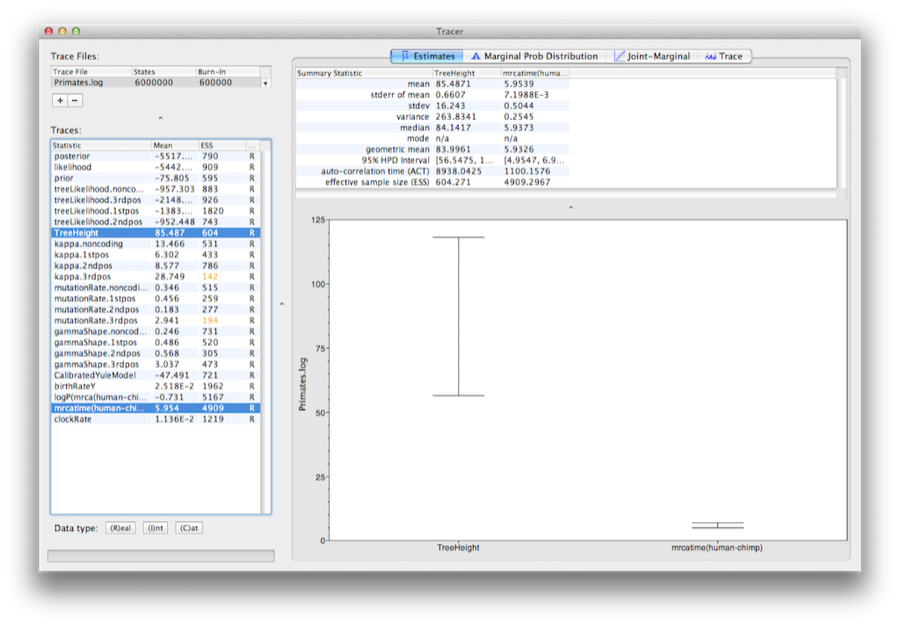
Figuur 14: Een screenshot van de 95% HPD-intervallen van de wortelhoogte en de door de gebruiker opgegeven (human-chimp) MRCA in Tracer.
5 Marginale posterieure schattingen
Om de relatieve percentages voor de vier partities weer te geven, selecteert u de parameter mutationRate voor elk van de vier partities, en selecteert u het tabblad marginale dichtheid in Tracer.Figuur 15 toont de marginale dichtheden voor de relatieve substitutiesnelheden. Uit de grafiek blijkt dat codonpositie 1 en 2 wezenlijk verschillende percentages hebben (0,456 tegenover 0,183) en beide veel langzamer zijn dan codonpositie 3 met een relatief percentage van 2,941. De niet-coderende partitie heeft een snelheid die het midden houdt tussen codonposities 1 en 2 (0,346). Alles bij elkaar suggereert dit resultaat sterke zuiverende selectie in zowel de coderende als niet-coderende regio’s van de uitlijning.
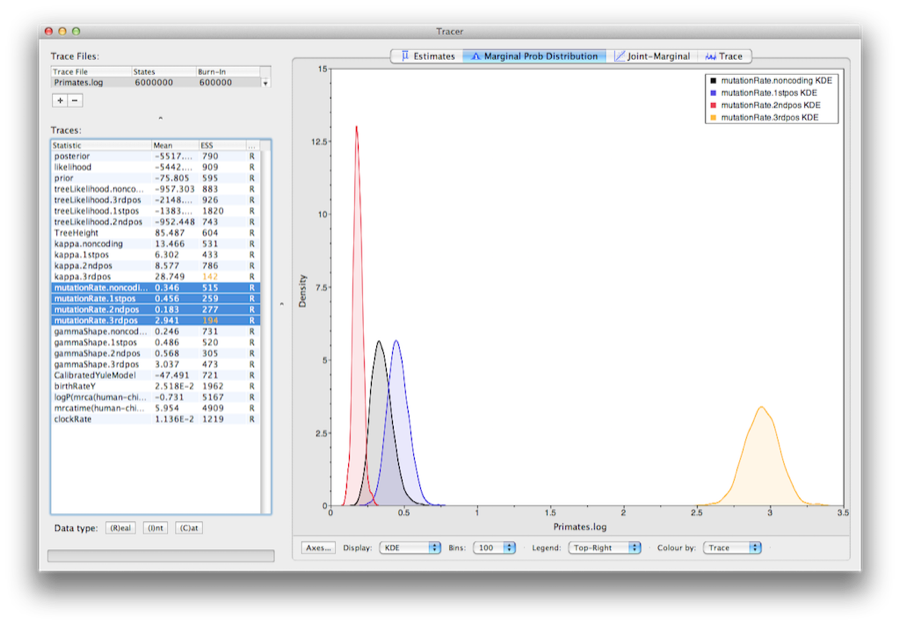
Figuur 15: Een screenshot van de marginale posterior dichtheden van de relatieve substitutiesnelheden van de vier partities (ten opzichte van de site-gewogen gemiddelde snelheid).
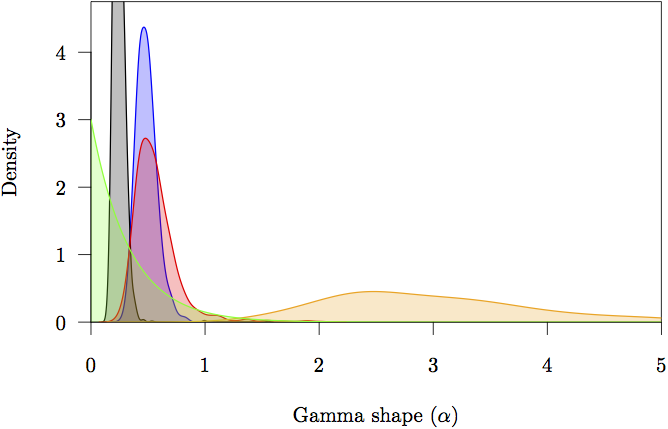
Figuur 16: De marginale prior en posterior densiteiten voor de vorm (α) parameters. De prior is in grijs weergegeven. De posterior dichtheidsschatting voor elke partitie wordt ook getoond: niet-coderende (oranje) en eerste (rode), tweede (groene) en derde (blauwe) codonposities.
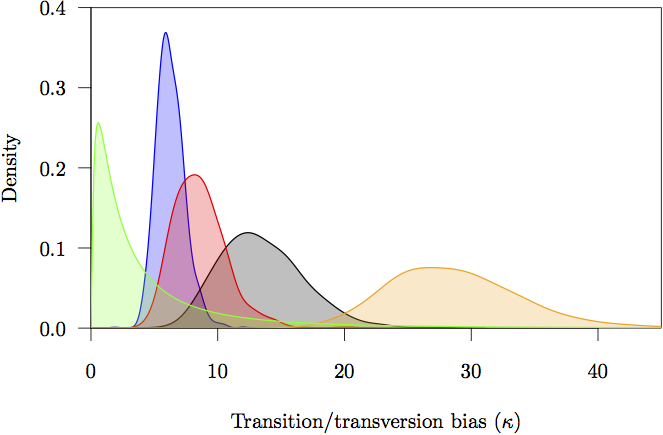
Figuur 17: De marginale prioriteits- en posterior dichtheden voor de overgangs-/transversiebias (κ)-parameters. De prior is in grijs. De posterior dichtheidsschatting voor elke partitie wordt ook getoond: niet-coderende (oranje) en eerste (rode), tweede (groene) en derde (blauwe) codonposities.
Vragen
Wat is de geschatte snelheid van moleculaire evolutie voor deze genboom (omvat het 95% HPD-interval)?
Welke foutenbronnen zijn in deze schatting opgenomen?
Hoe oud is de wortel van de boom (geef het gemiddelde en het 95% HPD-bereik)?
6 Het verkrijgen van een schatting van de fylogenetische boom
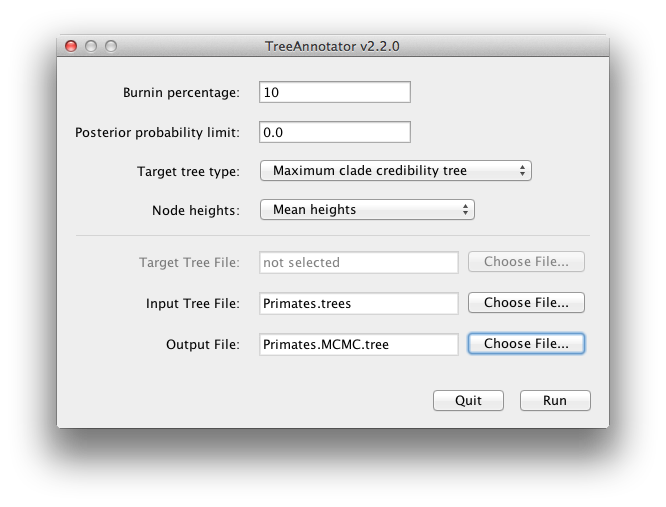
BEAST produceert ook een posterior monster van fylogenetische tijdbomen samen met zijn monster van parameterschattingen. Deze moeten worden samengevat met behulp van het programma TreeAnnotator. Dit zal de verzameling bomen nemen en de best ondersteunde vinden. Vervolgens annoteert het deze representatieve samenvattende boom met de gemiddelde leeftijden van alle thenoden en de bijbehorende 95% HPD-bereiken. Het berekent ook de posterior clade waarschijnlijkheid voor elk knooppunt. Start het programma TreeAnnotator en stel het in zoals afgebeeld in Figuur 18.
Figuur 18: Een screenshot van TreeAnnotator.
De burnin is het aantal bomen dat vanaf het begin van de steekproef moet worden verwijderd. In tegenstelling tot Tracer, dat het aantal stappen als burnin specificeert, moet je in TreeAnnotator het werkelijke aantal bomen specificeren. Voor deze run heb je een ketenlengte van 6.000.000 stappen gespecificeerd, waarbij elke 1.000 stappen worden bemonsterd. Het bomenbestand zal dus 6.000 bomen bevatten en dus moet je een 10% burnin opgeven in het bovenste tekstveld.
De optie Posterior probability limit specificeert een limiet zodat als een knooppunt met minder dan deze frequentie wordt gevonden in de steekproef van bomen (d.w.z. een posterior probability heeft die kleiner is dan deze limiet), het niet wordt geannoteerd. De standaardwaarde van 0.5 betekent dat alleen knooppunten die in de meerderheid van de bomen worden aangetroffen, worden geannoteerd. Stel dit in op nul om alle knooppunten te annoteren.
Het doeltype boom specificeert de boomtopologie die zal worden geannoteerd. U kunt een specifieke boom uit een bestand kiezen of TreeAnnotator vragen een boom in uw steekproef te vinden.De standaardoptie, Maximum clade credibility tree, vindt de boom met het hoogste product van de posterior waarschijnlijkheid van alle knooppunten.
Voor knoophoogten is de standaardoptie Common Ancestor Heights, die de hoogte van een knooppunt berekent als het gemiddelde van de MRCA-tijd van alle paren van knooppunten in de clade. Voor bomen met een grote onzekerheid in de topologie en dus veel clades met een lage ondersteuning kunnen sommige andere methoden resulteren in bomen met negatieve taklengtes. In deze analyse is de ondersteuning voor alle clades in de samenvattende boom zeer hoog, zodat dit hier geen probleem is.Kies Gemiddelde hoogtes voor knooppunthoogtes. Dit stelt de hoogtes (leeftijden) van elke knoop in de boom in op de gemiddelde hoogte over de gehele steekproef van bomen voor die clade.
Voor het invoerbestand selecteert u het bomenbestand dat BEAST heeft gemaakt en selecteert u een bestand voor de uitvoer (hier hebben we het Primates.MCC.tree genoemd). Druk nu op Uitvoeren en wacht tot het programma klaar is.
7 Visualiseren van de boomschatting
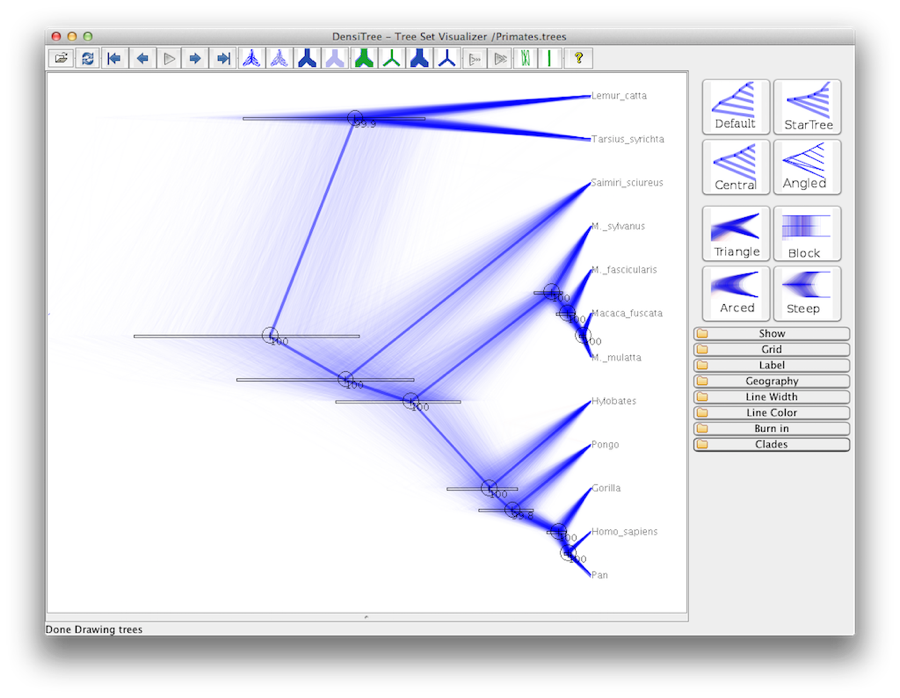
Tot slot kunnen we de boom visualiseren in een ander programma, FigTree genaamd. Start dit programma en open het bestand Primates.MCC.tree met de opdracht Open in het menu File. De boom zou moeten verschijnen. U kunt nu proberen enkele opties te selecteren in het configuratiescherm aan de linkerkant. Ten eerste, geef de optie Trees uit in het paneel, en vink Order nodes aan en kies Ordering by decreasing. Probeer Node Bars te selecteren om foutbalken voor de leeftijd van de knooppunten te krijgen. Zet ook Branch Labels aan en selecteer posterior om de posterior waarschijnlijkheid voor elke node te krijgen. Als u een niet strikt klokmodel gebruikt, kunt u onder Uiterlijk ook FigTree de opdracht geven om de takken te kleuren met de snelheid. U zou dan iets moeten krijgen dat lijkt op Figuur 19.
Figuur 19: Een screenshot van FigTree en DensiTree.
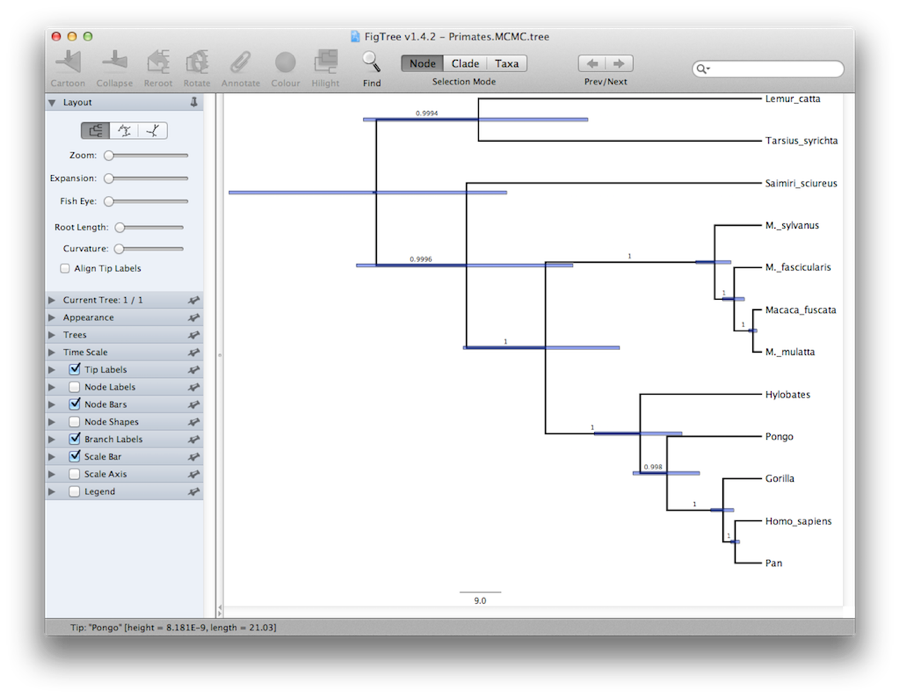
Een alternatieve weergave van de boom kan worden gemaakt met DensiTree, dat deel uitmaakt van Beast 2. Het voordeel van DensiTree is dat het zowel onzekerheid in knooppunthoogten als onzekerheid in topologie kan visualiseren.Voor deze specifieke dataset is de dominante topologie in meer dan 99% van de monsters aanwezig. We concluderen dus dat deze analyse resulteert in een zeer hoge consensus over de topologie (Figuur 19).
Vragen
- Verschilt de evolutiesnelheid aanzienlijk tussen de verschillende lineages in de boom?
- DensiTree heeft een cladebalk (Menu Window/View clade toolbar) om informatie over clades te tonen.
Wat is de ondersteuning voor de clade ?
- U kunt door de topologieën in DensiTree bladeren met behulp van het Bladeren menu.De meest populaire topologie heeft een ondersteuning van meer dan 99%.
Wat is de ondersteuning voor de op één na meest populaire topologie?
- Onder het help menu, toont DensiTree enige informatie.
Hoeveel topologieën zitten er in de boomverzameling?
8 Vergelijking van uw resultaten met de prior
Het is een goed idee om de analyse opnieuw uit te voeren terwijl u bemonstert uit de prior om er zeker van te zijn dat interacties tussen priors geen invloed hebben op uw prior-informatie. De interactie tussen priors kan problematisch zijn, vooral wanneer u kalibraties gebruikt, omdat dit betekent dat u meerdere priors op de boom moet zetten.
Zet met BEAUti dezelfde analyse op, maar selecteer onder de MCMC-opties de optie Sample from prior only. Hierdoor kunt u de volledige prioriteitsverdeling visualiseren bij afwezigheid van uw sequentiegegevens. Vat de bomen van de volledige prior verdeling samen en vergelijk de samenvatting met de posterior samenvatting boom.
Divergentie tijd schatting met behulp van “node dating” van het type beschreven in dit hoofdstuk is toegepast om een verscheidenheid van verschillende vragen in ecologie en evolutie te beantwoorden. Knooppuntdatering met fossielen werd bijvoorbeeld gebruikt bij het bepalen van de soortenrijkdom van cycaden, het analyseren van de evolutiesnelheid in bloeiende planten, en het onderzoeken van de oorsprong van cyanobacteriën in warme en koude woestijnen.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, e.a., Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond and Marc A Suchard, Bayesian random local clocks, or one rate to rule them all, BMC biology 8 (2010), no. 1, 114. A Gelman, G Roberts, and W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled and Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, and S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian, and Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith and Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, Science 322 (2008),no. 5898, 86-89.
Dit document is vertaald uit LATEX doorHEVEA.