Introduction
Germline de novo mutaties (DNMs) zijn genetische veranderingen in het individu veroorzaakt door mutagenese die optreedt in ouderlijke gameten tijdens de oogenese en spermatogenese. De term “de novo” moet hier niet verward worden met de term “nieuwe mutatie”. Hoewel DNM’s in de context van een trio (vader, moeder en kind) nieuwe mutaties zijn, kunnen zij in de algemene bevolking veel voorkomende, zeldzame of nieuwe varianten zijn. Om de snelheid van een bepaalde DNM te meten en te verklaren, is het nodig om eerst de impact van de variant op het fenotype te beoordelen, omdat nieuwe gunstige eigenschappen kunnen evolueren wanneer ontstane genetische mutaties een specifiek overlevingsvoordeel bieden (Front Line Genomics, 2017).
Bij mensen met genetische niet-Mendeliaanse ziekten die sporadisch voorkomen, zijn DNM’s meestal nieuw, betrouwbaarder en schadelijker dan overgeërfde varianten, omdat ze niet aan sterke natuurlijke selectie zijn onderworpen (Crow, 2000; Front Line Genomics, 2017). Daarom kan het identificeren van de genetische oorzaak van een door een DNM geïnduceerde stoornis in een individu vanuit klinisch oogpunt een uitdaging zijn, omdat pleiotropie en genetische heterogeniteit ten grondslag kunnen liggen aan een enkel fenotype (Eyre-Walker en Keightley, 2007). Dienovereenkomstig zijn er in het laatste decennium aanzienlijke inspanningen geleverd om exomen te sequencen van individuen met ziekten waarvan de genetische etiologie onduidelijk is, met het oog op klinische diagnostiek. Echter, zelfs na detectie van kandidaat de novo varianten, is er nog steeds onvoldoende informatie over de veel voorkomende en zeldzame varianten, wat een duidelijke conclusie over de pathogeniciteit van de geïdentificeerde de novo variant en zijn rol in ziekte uitsluit (Acuna-Hidalgo et al., 2016). Deze beperking kan worden verklaard door het feit dat de novo varianten meestal heterozygoot zijn en zowel extreem zeldzaam als veel voorkomend kunnen zijn. In gevallen van zeer zeldzame de novo varianten kan de pathogeniciteit van de variant moeilijk te bewijzen zijn, omdat er niet meer patiënten zijn met hetzelfde fenotype en dezelfde de novo variant. Bij veel voorkomende de novo varianten is het mogelijk dat de factoren die bepalend zijn voor de pathogeniciteit van de variant niet bekend zijn, met name als sommige individuen in de algemene bevolking de variant hebben maar niet de genetische ziekte hebben. Echter, ongeacht het percentage de novo varianten, kunnen beide typen varianten geschaald worden op basis van relatieve fitness en natuurlijke selectie.
De aangepastheid hangt af van vele factoren; daarom, om te beoordelen of een DNM pathogeen of adaptief is, en om te begrijpen waarom het met een bepaalde frequentie in de populatie voorkomt, is het nodig om de variant in geschikte omstandigheden te onderzoeken. Die omvatten milieu, leeftijd van de ouders, genomische context, epigenetica en andere factoren, omdat ze allemaal van invloed zijn op de waarde van de gemiddelde relatieve fitness die monotoon toeneemt, terwijl de sterkte van selectie afneemt (Peck en Waxman, 2018).
Het hoofddoel van deze studie was om de frequentie van voorkomende DNM’s op te helderen en om te bepalen hoe deze mutaties zijn verdeeld in de genomen van de algemene Litouwse bevolking. Wij hebben ook onderzocht of de frequentie van deze mutaties werd beïnvloed door de samenstelling of structurele parameters van de sequenties waarin zij voorkomen en door andere factoren die van invloed zouden kunnen zijn op de mechanismen die ten grondslag liggen aan de vorming van deze DNMs. Tenslotte hebben we getracht vast te stellen of DNM’s zijn ontstaan door intensieve druk van natuurlijke selectie op de functionele regio’s. Hoewel de verspreiding en intensiteit van DNM’s onderwerp zijn geweest van veel studies, waren ze nog niet eerder onderzocht in de Litouwse bevolking.
Materialen en Methoden
In deze studie analyseerden we monsters van de Litouwse bevolking, verkregen uit het LITGEN-project (LITGEN, 2011). De dataset bestond uit 49 trio’s met een totaal van 144 verschillende individuen. Genomisch DNA werd uit veneus bloed geëxtraheerd met behulp van de fenol-chloroform extractiemethode of het geautomatiseerde DNA-extractieplatform TECAN Freedom EVO® (Tecan Schweiz AG, Zwitserland) op basis van de paramagnetische deeltjesmethode. Exomen werden gesequenced op een SOLiD 5500 sequencing systeem (75 bp reads). De sequencing-gegevens werden verwerkt en geprepareerd met Lifescope-software. Exomen werden in kaart gebracht volgens het humane referentiegenoom build 19. De gemiddelde leesdiepte van sequencing was 38,5. BAM-geformateerde bestanden van moeder, vader en kind gegenereerd door Lifescope werden gecombineerd met behulp van SAMtools software voor elk trio.
De novo mutaties werden geïdentificeerd door twee softwareprogramma’s: VarScan (Koboldt et al., 2012) en VarSeqTM. Een potentiële variant werd geacht een DNM te zijn als deze werd geïdentificeerd in de nakomelingen, maar niet aanwezig was in een van de ouders op dezelfde positie. In totaal werden 1.752 en 4.756 DNMs gedetecteerd door respectievelijk VarScan en VarSeqTM. Om vals-positieve de novo calls te elimineren, wanneer het onbekend was of alle individuen in het trio correct waren geïdentificeerd, werden conservatieve filters op gedetecteerde DNM kwaliteitsparameters als volgt toegepast: (1) genotype kwaliteit van het individu ≥50; (2) aantal gelezen op elke site >20. SnpSift software werd gebruikt om deze filters toe te passen op de gegevens gegenereerd door VarScan. Gegevens gegenereerd door VarSeqTM software werden gefilterd door te kiezen voor dezelfde filtering parameters in de Trio Workflow segment. Bovendien, om de resterende varianten die somatisch waren (slechts aanwezig in een fractie van de gesequenced bloedcellen) met een laag allel evenwicht of sequencing artefacten te verwijderen, werden DNM’s gefilterd door het instellen van een drempel voor de waargenomen fractie van de leest in individuen met het alternatieve allel (het allel evenwicht) voor het trio (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Daarnaast werden alle mogelijk geïdentificeerde en gefilterde de novo single nucleotide varianten handmatig beoordeeld met Integrative Genomics Viewer (Robinson et al., 2011). Vanwege het grote aantal geïdentificeerde DNMs, werden voor de validatie van varianten door Sanger sequencing, 51 de novo single nucleotide varianten willekeurig geselecteerd. Sanger sequencing werd uitgevoerd met behulp van een ABI PRISM 3130xl Genetic Analyzer. Alle gefilterde en handmatig beoordeelde DNM’s geïdentificeerd door VarScan (N = 95) en door VarSeqTM (N = 84) werden geannoteerd met behulp van ANNOVAR (Butkiewicz and Bush, 2016; Wang et al., 2010). Voor de analyse van interacties tussen eiwitten werd gebruik gemaakt van STRING software (Szklarczyk et al., 2017). Net als in het geval van exoomkartering werden annotaties uitgevoerd met behulp van hg19 referentie menselijk genoom.
De waarschijnlijkheid dat een aanroepende positie een DNM in het trio was, werd voor elk trio onafhankelijk berekend. Zoals beschreven in een eerdere referentie (Besenbacher et al., 2015), werd de de novo rate per positie per generatie (PPPG) als volgt berekend:
waarbij f het aantal trio’s is en N het aantal opvraagbare sites, dat potentieel kan worden geïdentificeerd als de novo sites voor elk trio afzonderlijk, ongeacht de sequencing diepte. Dit aantal varieert naar gelang van het trio. ni is het aantal geïdentificeerde DNM’s voor trio i. De kans dat Pji (de novo ingle nucleotide) voor de opgeroepen single nucleotide site j en familie i gemuteerd is, werd als volgt berekend
De kans Pji (de novo indel) voor de opgeroepen indelplaats j en familie i om gemuteerd te zijn, werd berekend als:
waar C, M, en F respectievelijk staan voor nakomeling, moeder, en vader, en Hetero, HomR, en HomA respectievelijk heterozygoot, homozygoot voor referentie, en homozygoot voor alternatief allel aanduiden. De waarschijnlijkheid Pij (de novo) werd berekend ten opzichte van de sequencing-dekking. Betrouwbaarheidsintervallen voor de kansschattingen werden berekend zoals voor binomiale proporties. Voor de schatting van de DNM rate en voor verdere berekeningen gebruikten we het R-pakket (versie 3.4.3) (R Core Team, 2013).
Om de hypothese te testen dat variaties in DNM-snelheid in verschillende regio’s van het genoom kunnen worden verklaard door intrinsieke kenmerken van de genomische regio zelf en de leeftijd van de ouder, werd lineaire regressieanalyse uitgevoerd, waarvoor de “secundaire” annotatie van elke DNM werd uitgevoerd met behulp van gegevens uit ENCODE (ENCODE Project Consortium, 2012) en LITGEN (LITGEN, 2011) projecten. Ten eerste, volgens een eerdere studie (Besenbacher et al., 2015), om gegevens te verzamelen met betrekking tot het genomische landschap van de geïdentificeerde DNMs, werden lymfoblastoïde cellijnen (LCL en GM12878) (ENCODE Project Consortium, 2012) gekozen. Gegevens werden verzameld voor:
(1) expressiepercentages (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) in verschillende weefsels. Volgens expressie van regio’s met DNMs werden verdeeld in posities met specifieke en niet-specifieke expressie;
(2) metingen van DNase1 overgevoeligheid sites (DHS). DHS-status werd toegekend 0 indien buiten DHS-piek en 1 indien binnen;
(3) metingen van de context van CpG-eilanden. Als DNM zich binnen CpG-eilanden bevond, werd een positiestatus toegekend van 1; indien erbuiten – 0;
(4) drie histon-markeringen (H3K27ac, H3K4me1, en H3K4me3) van het ENCODE-project. Als DNM zich op een met histon gemarkeerde positie bevond, werd er een 1 aan toegekend en zo niet – 0;
(5) GERPP++ behoudswaarden werden verzameld met behulp van ANNOVAR annotatie-tool. Volgens de behoudswaarden werden posities met DNM’s ingedeeld in conservatieve (GERP++ score >12) en niet-conservatieve posities (GERP++ score <12) (Davydov et al., 2010; ENCODE Project Consortium, 2012). Op basis van vragenlijstgegevens van het LITGEN-project werden gegevens over de leeftijd van de ouders verzameld. Na het verzamelen van parameters voor elk trio werd een aantal posities met elke parameter berekend. Vervolgens werd een correlatie-analyse gevolgd door lineaire regressie modellering van DNM percentage en parameters uitgevoerd.
Resultaten
Na DNM analyse werd een uitzonderlijk hoog aantal DNMs geïdentificeerd voor twee trio’s (nrs. 4 en 21): 113 en 123 (door VarScan en VarSeqTM, respectievelijk) en 16 (VarScan). Deze bevindingen zetten ons ertoe aan het biologisch vaderschap te testen, wat werd verworpen voor trio nr. 4 en bevestigd voor trio nr. 21. Daarom werden de gegevens van trio nr. 4 van de studie uitgesloten. In de uiteindelijke set van 48 trio’s werden 95 DNM’s geïdentificeerd in 34 trio’s met VarScan software en 84 DNM’s in 31 trio’s werden geïdentificeerd met VarSeqTM software (Figuur 1). Er werden geen DNM’s gedetecteerd in 18 en 15 trio’s door respectievelijk VarScan en VarSeqTM. Van alle DNM’s die door beide softwareprogramma’s werden geïdentificeerd, matchte slechts 5,37% (drie DNM’s in MEIS2-, PGK1- en MT1B-genen). Elke persoon had gemiddeld 1,9 (VarScan-software) en 1,7 (VarSeqTM) DNM’s.
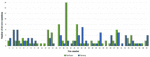
FIGUUR 1. Vergelijking van de novo single nucleotide varianten geïdentificeerd door VarScan (blauw) en VarSeqTM (groen) software.
Analyse van 95 DNMs die werden geïdentificeerd door VarScan software toonde aan dat 20 DNMs exonisch waren, waaronder twee stop-gain DNMs, zeven synonieme DNMs, en 11 niet-synonieme DNMs. Tachtig nieuwe mutaties geïdentificeerd door VarSeqTM waren exonisch, waaronder 1 stop-gain DNM en 78 niet-synonieme DNMs (figuur 2). De meerderheid van DNMs geïdentificeerd door VarScan waren in chromosomen 1, 2, 4, en 5, terwijl VarSeqTM DNMs identificeerde voornamelijk in chromosomen 2, 6, 7, en 11. Het aantal geïdentificeerde DNMs niet correleren met de dichtheid van genen in de chromosomen (R = 0,09, p-waarde = 0,65 voor VarScan en R = 6,73, p-waarde = 0,51 voor VarSeqTM) of met de chromosoom grootte (figuur 3). Volgens beide softwareprogramma’s waren de verhoudingen van overgangen en transversies zeer vergelijkbaar: 1,44 en 1,47, respectievelijk (figuur 4). Er werden echter verschillen in de structuren van de overgangen vastgesteld. Meer bepaald waren er bij de met VarScan geïdentificeerde DNM’s meer G/T- en A/C-veranderingen, terwijl er bij de met VarSeqTM geïdentificeerde DNM’s meer A/T- en G/C-veranderingen waren.
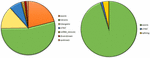
FIGUUR 2. De samenstelling van de novo mutaties (DNM’s) gegenereerd door VarScan (links) en door VarSeqTM (rechts).
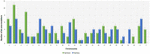
FIGUUR 3. Verdeling van het aantal de novo varianten per chromosoom volgens de VarScan en VarSeqTM gegenereerde gegevens. Groene balken vertegenwoordigen DNMs geïdentificeerd door VarScan software, blauw – door VarSeqTM. De foutbalken vertegenwoordigen de standaardfout van de gemiddelde DNMs voor elk chromosoom.
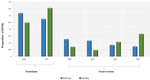
FIGUUR 4. De moleculaire gebeurtenissen die ten grondslag liggen aan overgangen komen vaker voor dan die welke leiden tot transversies, wat resulteert in een ∼1,5-voudig hoger percentage van overgangen boven transversies over het hele exoom. Overgangs- en transversiegebeurtenissen geïdentificeerd door VarScan (groen) en VarSeqTM (blauw) software. De foutbalkjes geven de standaardfout van de gemiddelde DNM’s weer.
De berekende percentages de novo single nucleotide mutaties waren 2,4 × 10-8 PPPG (95% betrouwbaarheidsinterval : 1,96 × 10-8-2,99 × 10-8) volgens VarSeqTM en 2.74 × 10-8 per nucleotide per generatie (95% CI: 2,24 × 10-8-3,35 × 10-8) volgens VarScan.
Drie de novo indels in drie trio’s werden geïdentificeerd door het VarScan-algoritme in chromosomen 6 en 11. Het berekende percentage de novo indels in het genoom was 1,77 × 10-8 (95% CI: 6,03 × 10-9-5,2 × 10-8) PPPG. Opmerkelijk is dat alle de novo indels “omkeerbaar” waren, d.w.z. dat de ouders nieuwe varianten in het genoom hadden, en hun kinderen de novo varianten op basis van het referentiegenoom met respectievelijk de gemiddelde waarde van 37,5 voor de diepte van de sequencing en 50 voor de genotypekwaliteit. Deze drie DNM’s werden echter niet geselecteerd voor de validatie door Sanger sequencing methode waardoor de kans op overschatting van de novo indels niettemin blijft bestaan. De novo indels waren C/T en A/G in de context van enkele nucleotiden.
Lineaire regressiemodellering toonde aan dat DNAse 1 overgevoeligheidsplaatsen, context van CpG eilanden, GERPP++ behoudswaarden, en expressieniveaus ∼68-93% van DNM percentages verklaarden (Tabel 1). Noch epigenetische markers, noch de vaderlijke leeftijd correleerden significant met het DNM percentage. De modellen werden uitsluitend op basis van de gegevens van VarScan opgesteld omdat er geen correlatie was tussen gegevens van VarSeqTM en intrinsieke kenmerken van het genoomgebied zelf.

TABLE 1. De lineaire regressie van de DNAaseI overgevoeligheid sites, context van CpG eilanden, GERPP++ behoud waarden en expressie niveau effect van op de snelheid van DNMs.
Functionele voorspelling van DNMs
Om te beoordelen welke missense mutaties waren deleterious en veranderde de functie van het getroffen eiwit per type, voorspelde categorische scores voor de schade geïnduceerd door DNMs werden geanalyseerd. De volgende 10 waarden werden in aanmerking genomen: polyphen HDIV en HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, Fathmm-MKL-codering, en GERP++. Op basis van de voorspelde scores werden vier DNM’s geselecteerd die volgens VarScan zes of meer schadelijke of waarschijnlijk schadelijke voorspellingen hadden. Deze stop-gain DNMs bevonden zich in de MEIS2 en ULK4 genen, terwijl niet-synonieme DNMs zich in de MT1B en PGK1 genen bevonden. Eiwitten gecodeerd door deze genen zijn belangrijk voor neuronale groei, endocytose, en bescherming tegen de negatieve effecten van zware metalen. Deze eiwitten nemen deel aan het vrijkomen van de tumorbloedvatremmer angiostatine en aan verschillende signaalwegen. Er waren geen verbanden tussen de door deze genen gecodeerde eiwitten (figuur 5).
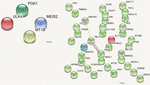
FIGUUR 5. Eiwit-eiwit interacties (Szklarczyk et al., 2017) in genen die DNM’s herbergen. DNM’s geïdentificeerd door VarScan in genen die coderen voor eiwitten staan links, DNM’s geïdentificeerd door VarSeqTM – rechts. Gekleurde lijnen geven een verband aan tussen eiwitten.
De novo mutaties geïdentificeerd door VarSeqTM werden in meer detail geanalyseerd als ze door ten minste de helft van de voorspellingstools als schadelijk of waarschijnlijk schadelijk werden voorspeld. Er waren 35 puntmutaties (zie ??) in genen die coderen voor eiwitten die belangrijk zijn voor chromatine remodellering, regulering van het cytoskelet, celgroei en levensvatbaarheid, cytoplasmatische signaalwegen, en de initiatie van neuronale reacties die de perceptie van geur in gang zetten.
Van de eiwitten die werden gecodeerd door de DNM-beïnvloede genen, waren alleen CLPTM1, ZNF547 en DMXL1 op de een of andere manier met elkaar verbonden (figuur 5).
Discussie
In deze studie voerden we een uitgebreide analyse uit van de verdeling van DNM’s over verschillende regio’s van het exoom in de Litouwse bevolking. In totaal werden 95 DNM’s in 34 trio’s en 84 DNM’s in 31 trio’s gedetecteerd met behulp van SOLiD 5500 sequencing technologie door respectievelijk VarScan en VarSeqTM algoritmen. Eerst en vooral willen we opmerken dat we VarScan kozen voor het oproepen van DNMs omdat dit algoritme volgens (Warden et al., 2014) een lijst van varianten oplevert, met een hoge concordantie (>97%) met varianten van hoge kwaliteit opgeroepen door de GATK UnifiedGenotyper en HaplotypeCaller. VarSeqTM software werd gekozen omdat het een veelgebruikt instrument is voor varianten analyse, zowel in onderzoek als klinische analyse. Ondanks het feit dat beide algoritmen ontworpen zijn om te zoeken naar DNMs in het exoom van het nageslacht die niet aanwezig waren in één van beide ouders, was de overeenkomst tussen de twee softwareprogramma’s voor DNM analyse slechts 5.37%. Het VarScan algoritme had een hogere gevoeligheid (5.42%) voor DNM detectie vóór filtratie dan het VarSeqTM algoritme (1.77%). We vermoedden dus dat geen enkel instrument erin slaagde mutaties op te roepen vanwege een hoge gevoeligheid die altijd gepaard ging met een lage specificiteit. Daarom suggereren we dat een aanzienlijke verbetering van de resultaten zou kunnen worden bereikt door de output van verschillende tools te combineren (Sandmann et al., 2017).
Op basis van de gegenereerde gegevens was de geschatte single nucleotide DNM rate tussen 2,4 × 10-8 en 2,74 × 10-8 en die van de novo indels was 1,77 × 10-8 PPPG, afhankelijk van het gebruikte algoritme. Onze berekende DNM rate was hoger dan die gerapporteerd in eerdere studies (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015), waarin het varieerde tussen 1,2 × 10-8 en 1,5 × 10-8 PPPG. De hogere DNM rate in onze studie was redelijk omdat onze studie gebaseerd was op exoomgegevens. Bovendien vertonen exomen significant hogere (30%) mutatiepercentages dan volledige genomen omdat de basenpaar-samenstelling van het volledige genoom anders is dan die van exomen. In het bijzonder hebben exomen een gemiddeld GC-gehalte van ongeveer 50%, terwijl dat van het volledige genoom ongeveer 40% is (Neale et al., 2012). Gemethyleerde CpG’s vertegenwoordigen zeer mutabele sequenties bij de mens als gevolg van de spontane deaminatie van cytosine basen (Neale et al., 2012). Volgens vergelijkende genomics-studies zouden de verhoogde mutatiepercentages in CpG-rijke regio’s zijn geëvolueerd rond de tijd van de zoogdierstraling (Francioli et al., 2015). Tijdens de divergentie van soorten ondergingen CpG-rijke exonische regio’s verhoogde mutatiesnelheden in vergelijking met die in niet-coderend DNA en veranderden in niet-coderende regio’s. Daarom, dan het effect van CpG-gehalte afnemen in de tijd, de gemiddelde snelheid van mutatie dalen tot het niveau aanwezig in de omliggende niet-coderende DNA (Subramanian en Kumar, 2003). Echter, terwijl sequenties in neutraal evoluerende regio’s van het genoom voldoende tijd hebben gehad om te equilibreren met betrekking tot dinucleotide contexten, heeft zuiverende selectie hypermutabele CpGs in functionele regio’s in stand gehouden (Subramanian en Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Omdat wij een hoger DNM-percentage vonden dan in andere studies werd gerapporteerd, speculeerden wij daarom dat dit ten minste gedeeltelijk te wijten zou kunnen zijn aan de lokale sequentiecontext en/of mogelijke natuurlijke selectiedruk op het exoom. Dienovereenkomstig werd een lineair regressiemodel toegepast, en wij vonden dat DNAse 1 overgevoeligheid, context van CpG-eilanden, GERPP++ behoudswaarden, en expressieniveau ∼68-93% van de DNM rate verklaarden. Deze bevindingen wezen erop dat DNM’s in het exoom onafhankelijk van het behoud van DNA-sequenties werden gevormd. De DNM rate was echter hoger in genen waarvan de producten niet-specifiek waren en in transcriptioneel actieve promotor-achtige regio’s.
In tegenstelling tot de resultaten van andere studies (Wong et al., 2016; Sandmann et al., 2017), vonden we dat de vaderlijke leeftijd niet correleerde met de DNM rate. Deze bevindingen zouden kunnen worden verklaard door het feit dat de dataset bestond uit trio’s met vergelijkbare ouderleeftijden en dat slechts een klein deel (∼1,5%) van het volledige genoom werd geanalyseerd. Op basis van deze parameters had elke persoon gemiddeld slechts 1,9 (VarScan) of 1,7 (VarSeqTM) DNM’s, vergeleken met 40-82 in het volledige genoom (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al, 2016), terwijl het aantal de novo indels in de coderende sequentie vergelijkbaar was met die geïdentificeerd in (Front Line Genomics, 2017).
De resultaten van onze uitgebreide functionele analyse van annotaties toonden aan dat van alle geïdentificeerde DNM’s, 4 (VarScan) en 35 (VarSeqTM) varianten waarschijnlijk pathogene DNM’s waren. Het verschil in het aantal pathogene DNM’s kan verklaard worden door het feit dat afhankelijk van het algoritme dat gebruikt werd voor de identificatie van DNM’s, het aandeel van DNM’s in coderende sequenties aanzienlijk verschilde. Zo was 21,05% van de DNM’s die door de VarScan-software werden geïdentificeerd exonisch, terwijl 95,24% van de DNM’s die door de VarSeqTM -software werden geïdentificeerd exonisch waren. Deze pathogene DNM’s zaten in de genen die coderen voor eiwitten die essentieel zijn voor chromatine modellering, regulering van het cytoskelet, modulatie van celgroei en vitaliteit, functie van cytoplasmatische signaalwegen, en initiatie van neuronale respons. Hoewel deze DNM’s als pathogeen worden beschouwd, identificeerden alle deelnemers aan de enquête zich als genetisch “gezond”. Dit resultaat wees er dus op dat ondanks de vermeende pathogeniteit van DNM’s, de genomen waarin DNM’s zich bevonden dergelijke veranderingen duidelijk tolereerden, zodat ziekteverschijnselen vaak niet uitgesproken waren. Volgens Szamecz et al. (2014) zijn de effecten van natuurlijke selectie op genetische veranderingen door compenserende mechanismen van genoombescherming sterker naarmate DNM’s vaker voorkomen op geconserveerde genetische posities. De schadelijke effecten van de varianten kunnen op vier manieren worden gemitigeerd. Sommige genen kunnen afgekapte varianten van eiwitten tolereren, omdat hun functionele effecten worden gemaskeerd door onvolledige expressie, compenserende varianten, of een lage functionele betekenis van de truncatie (Bartha et al., 2015). Daarentegen worden genveranderingen geassocieerd met niet-synonieme DNM’s gecompenseerd via het mechanisme van nuttige mutatie-accumulatie in het hele genoom (Szamecz et al., 2014). Dit suggereert dat in deze gevallen de pathogene mutaties niet deleterieus genoeg zijn om de gemiddelde fitness te verminderen en daarom blijven ze langer bestaan in vele generaties die door natuurlijke selectie worden gevormd.
Samenvattend heeft onze analyse van de verspreiding van DNM’s en van hun genetische en epigenetische context inzichten verschaft in de genetische variatie van het Litouwse genoom. Op basis van deze bevindingen kunnen aanvullende studies bij patiëntengroepen met genetische ziekten ons vermogen om bepaalde pathogene DNM’s van de getolereerde achtergrond-DNM’s te onderscheiden en betrouwbare oorzakelijke DNM’s te identificeren, vergemakkelijken. De belangrijkste beperking van deze studie was echter dat we de variatie in niet-coderende en regulerende genregio’s niet hebben onderzocht. Deze informatie zou kunnen bijdragen tot de opheldering van mogelijke mechanismen van DNM-vorming die nog onvoldoende duidelijk zijn.
Accessiecodes
De sequentiegegevens zijn gedeponeerd bij het European Nucleotide Archive (ENA), onder toetreding PRJEB25864 (ERP107829).
Ethics Statement
Deze studie werd uitgevoerd in overeenstemming met de aanbevelingen van de toestemming, Vilnius Regionale Ethische Commissie voor Biomedisch Onderzoek. Het protocol werd goedgekeurd door het Vilnius Regionaal Ethisch Comité voor Biomedisch Onderzoek. Alle proefpersonen gaven schriftelijke geïnformeerde toestemming in overeenstemming met de Verklaring van Helsinki.
Author Contributions
LP voerde de gegevensanalyse uit en stelde het manuscript op. AJ berekende het percentage de novo mutaties. Sequencing van trios exomen werd uitgevoerd door LA en IK. VK was de hoofdonderzoeker.
Funding
Deze studie werd ondersteund door het Europees Sociaal Fonds in het kader van de Global Grant maatregel. LITGEN project nr. VP1-3.1-ŠMM-07-K-01-013.
Conflict of Interest Statement
De auteurs verklaren dat het onderzoek is uitgevoerd in afwezigheid van enige commerciële of financiële relaties die zouden kunnen worden opgevat als een potentieel belangenconflict.
Supplementary Material
Het Supplementary Material voor dit artikel is online te vinden op: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). Nieuwe inzichten in het ontstaan en de rol van de novo mutaties in gezondheid en ziekte. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). De kenmerken van heterozygote eiwit truncerende varianten in het menselijk genoom. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). In silico functionele annotatie van genomische variatie. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). De oorsprong, patronen en implicaties van spontane mutatie bij de mens. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identificatie van een hoge fractie van het menselijk genoom onder selectieve beperking met behulp van GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Een geïntegreerde encyclopedie van DNA-elementen in het menselijk genoom. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | PubRef Full Text | Google Scholar
Eyre-Walker, A., and Keightley, P. D. (2007). The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genoomwijde patronen en eigenschappen van de novo mutaties bij de mens. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics Magazine Issue 14 – ASHG. Londen: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Genetische effecten op genexpressie in menselijke weefsels. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: ontdekking van somatische mutatie en kopiegetalverandering in kanker door exoomsequencing. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Fijnschalige verschillen in recombinatiesnelheid tussen geslachten, populaties en individuen. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ’t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Transcriptoom- en genoomsequencing leggen functionele variatie bloot in de mens. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Beschikbaar op: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patronen en percentages van exonische de novo mutaties in autismespectrumstoornissen. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | PubMed Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Wat is adaptatie en hoe moet het gemeten worden? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). Een taal en omgeving voor statistische berekeningen. Wenen: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integratieve genomics viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., and Sunyaev, S. (2008). Hypermuteerbare niet-synonieme sites staan onder sterkere negatieve selectie. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Neutrale substituties komen in een sneller tempo voor in exonen dan in niet-coderend DNA in genomen van primaten. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). Het genomische landschap van compensatoire evolutie Be. Het genomische landschap van compensatoire evolutie. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). De STRING database in 2017: kwaliteitsgecontroleerde eiwit-eiwit associatie netwerken, breed toegankelijk gemaakt. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functionele annotatie van genetische varianten uit next-generation sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Gedetailleerde vergelijking van twee populaire variant calling pakketten voor exoom en gerichte exon studies. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). New observations on maternal age effect on germline de novo mutations. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar
.