John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Growing evidence highlights the intimate relationship between type II diabetes (T2D) and Alzheimer’s disease (AD). Co ważne, te dwie choroby wykazują wiele podobieństw patologicznych, w tym akumulację amyloidu, stres oksydacyjny, zapalenie i śmierć komórek. Do tej pory brakuje leków na AD i T2D i istnieje ogromna potrzeba odkrycia i opracowania nowych terapii dla tych chorób. W wielu badaniach na ludziach i gryzoniach udowodniono, że suplementacja hormonów metabolicznych jest bardzo cenna dla poprawy funkcji poznawczych i ogólnego stanu metabolicznego zarówno w T2D jak i AD. Hormon trzustkowy amylina stał się kluczowym elementem etiologii zarówno T2D jak i AD, chociaż dokładna rola amyliny w tych chorobach nie jest jeszcze dobrze poznana. W tym miejscu dokonano krytycznego przeglądu aktualnej literatury wykorzystującej ludzką amylinę lub jej syntetyczny analog, pramlintyd, jak również antagonistów receptora amylinowego w leczeniu AD.
Wprowadzenie
Choroba Alzheimera (AD) jest postępującą, osłabiającą chorobą neurodegeneracyjną charakteryzującą się gromadzeniem blaszek amyloidu-beta (Aβ) i splątań neurofibrylarnych złożonych z hiperfosforylowanego tau1. Nagromadzenie tych patologicznych peptydów przyczynia się do deficytów funkcji wykonawczych, takich jak uczenie się i pamięć, nastrój, afekt, itp. oraz stanowi znaczne obciążenie dla pacjenta i jego opiekunów. Częstość występowania AD rośnie w alarmującym tempie w Stanach Zjednoczonych, szacuje się, że w 2017 r. z AD żyło 5,5 mln Amerykanów, a liczba ta ma się potroić do 2050 r.2. Co więcej, koszty opieki i leczenia pacjentów z AD przekraczają obecnie 200 miliardów dolarów rocznie i oczekuje się, że będą rosły3. Chociaż choroba Alzheimera jest niewątpliwie ogromnym problemem w Stanach Zjednoczonych i poza nimi, możliwości leczenia pozostają bardzo ograniczone4. Przeprowadzono wiele badań nad lekami o różnym przeznaczeniu, ale obecnie FDA zatwierdziła tylko sześć leków na AD i są to jedynie terapie objawowe5, 6. Do tej pory większość opracowanych środków farmakologicznych była ukierunkowana na patologię Aβ lub tau, ale żaden z nich nie okazał się skuteczny w usuwaniu patologii lub zapobieganiu jej4. W związku z tym istnieje fundamentalna potrzeba opracowania skutecznych terapii i metod zapobiegania chorobie Alzheimera.
Sporadikowa choroba Alzheimera związana z wiekiem jest skomplikowaną, wieloczynnikową chorobą o licznych wpływach genetycznych i środowiskowych. Środowisko i styl życia są silnie związane z rozwojem sporadycznej AD; czynniki takie jak dieta7-9, otyłość8-10, zespół metaboliczny7, cukrzyca typu II (T2D)9, 11 i choroby sercowo-naczyniowe12 zostały włączone do przyczyn AD. Co niezwykle istotne, częstość występowania otyłości i cukrzycy gwałtownie rośnie równolegle z występowaniem choroby Alzheimera12, 13. Chociaż związek między otyłością a chorobą Alzheimera jest niejasny, istnieją dowody na to, że otyłość w średnim wieku odgrywa rolę w rozwoju choroby Alzheimera10. Co ważniejsze, otyłości powszechnie towarzyszy wiele innych chorób, w tym choroby układu krążenia, nadciśnienie tętnicze, dyslipidemia, T2D, udar mózgu itd.14. Częstość występowania T2D szybko rośnie, CDC szacuje, że około 30,3 miliona osób (1 na 10 dorosłych) w USA ma cukrzycę, a oszałamiające 84,1 miliona (1 na 3 dorosłych) ma prediabetes, przy czym większość z nich nie jest świadoma swojego stanu. Ponadto, ze względu na zmniejszenie aktywności fizycznej na dużą skalę, któremu towarzyszy jednoczesny wzrost spożycia żywności i nieprawidłowa dieta, szacuje się, że wskaźniki otyłości, T2D, zespołu metabolicznego i chorób sercowo-naczyniowych wzrosną do około 600 milionów przypadków T2D na całym świecie do 2035 roku15.
Zasób dowodów wskazujących na udział funkcji metabolicznych i chorób w procesie pogorszenia funkcji poznawczych i starzenia się jest znaczący16, 17. Na przykład około 70% osób, u których rozpoznano T2D, zgłasza zaburzenia funkcji poznawczych, a u znacznej liczby pacjentów z T2D w późniejszym okresie rozwija się demencja16, 18-21. Osoby, u których T2D rozpoznaje się od co najmniej pięciu lat, mają znacznie zwiększone ryzyko rozwoju AD w porównaniu z osobami, które cierpią na T2D od mniej niż pięciu lat17. Łącznie dane te sugerują, że coraz częstsze występowanie T2D w populacji może przyczyniać się do wzrostu częstości występowania AD.
T2D początkowo charakteryzuje się wysokim stężeniem glukozy i insuliny we krwi, co prowadzi do hiperinsulinemii; co ważne, amylina, niewielki hormon metaboliczny produkowany przez komórki β-wysp trzustkowych, jest współpakowana i współwydzielana z insuliną, a zatem jest nadprodukowana w T2D.22. Co ważne, istnieje szereg cech patologicznych, które są obecne zarówno w T2D, jak i AD: 1) zmniejszony metabolizm mózgu i oporność na hormony metaboliczne 2) patologia amyloidowa 3) stres oksydacyjny (OS) i stan zapalny. Przewlekła hiperinsulinemia i hiperamylinemia prowadzi do szeregu problemów fizjologicznych: przewlekła hiperinsulinemia prowadzi do insulinooporności ustroju22, upośledzenia transportu insuliny przez barierę krew-mózg (BBB)23, 24, a tym samym do zmniejszenia sygnalizacji insulinowej w mózgu25. Utrata sygnalizacji insulinowej w mózgu wiąże się z szeregiem cech patologicznych związanych z chorobą Alzheimera, w tym ze zwiększoną produkcją Aβ, fosforylacją tau i neurozapaleniem.
Co więcej, amylina wykazuje podobne cechy patologiczne jak Aβ w wysokich stężeniach26 i może być wspólną ścieżką między tymi dwiema chorobami. Na przykład, fibryle amyliny znaleziono w trzustce 95% pacjentów z T2D27-29 i powodują one szereg zaburzeń fizjologicznych, w tym nieprawidłowy napływ Ca2+, zwiększone wydzielanie cytokin prozapalnych30,31 i ostatecznie utratę komórek β-trzustki32. Ponadto amylina łatwo przekracza BBB i tworzy fibryle amylinowe, jak również blaszki mieszane z Aβ w mózgu i może być odpowiedzialna za patologię podobną do AD i wysiew Aβ w T2D33-35. Wiadomo, że amylina wpływa na potencjalizację długoterminową (LTP) w hipokampie i może mieć wrodzony wpływ na funkcje poznawcze w mózgu36-39. Jednak to, czy amylina jest toksycznym czynnikiem uszkadzającym w tych chorobach lub czy jej utrata funkcjonalna poprzez agregację lub utratę komórek β w późnym stadium T2D przyczynia się do rozwoju AD, pozostaje niejasne.
Dychotomia sygnalizacji amylinowej
Wciąż toczy się wiele dyskusji na temat udziału receptora amylinowego (AMYR) i sygnalizacji amylinowej w progresji choroby i etiologii T2D i AD. Liczba badań mających na celu ustalenie tego związku szybko się powiększa. Wszystkie istotne badania konsekwentnie wykazują, że modulacja sygnalizacji amylinowej wpływa na patologię związaną z AD. Natura tego związku nie została jednak jeszcze dokładnie wyjaśniona. Kilka grup przedstawiło przekonujące dane sugerujące, że sygnalizacja amylinowa jest korzystna w zapobieganiu patologii związanej z AD i deficytom poznawczym zarówno in vivo, jak i in vitro40-44. Co ważne, pramlintid, rekombinowana nieagregująca postać amyliny, stosowana w połączeniu z terapiami insulinowymi w leczeniu cukrzycy i poprawiająca kontrolę glikemii, zmniejszająca masę ciała i zmniejszająca stężenie markerów OS w surowicy45-47 , jest również obiecująca jako terapia AD. Do tej pory nie przeprowadzono jednak badań klinicznych, których celem byłoby wykorzystanie amyliny lub pramlintydu jako środka terapeutycznego w leczeniu otępienia. Wyraźne dowody z badań na gryzoniach sugerują, że przewlekłe leczenie ludzką amyliną lub pramlintydem przynosi silne korzyści terapeutyczne w zmniejszaniu patologii związanej z AD; suplementacja amyliną/pramlintydem zmniejsza poziom rozpuszczalnego Aβ, obciążenie blaszkami miażdżycowymi, fosforylację tau, neurozapalenie i OS, jednocześnie poprawiając zdolności poznawcze40-42,44. Powyższe dane sugerują, że utrata wrodzonej sygnalizacji amylinowej w OUN z powodu agregacji daje zwiększone ryzyko rozwoju AD i jest omówiona bardziej szczegółowo w Grizzanti i wsp. 201848.
W przeciwieństwie do tego, badania pokazują również, że ludzka amylina i Aβ mają podobne działanie toksyczne i że te toksyczne efekty mogą być złagodzone przy użyciu antagonisty AMYR36-39,49. Na przykład, dane pokazują, że leczenie in vivo antagonistami AMYR przynosi bardzo podobne korzyści fizjologiczne jak leczenie amyliną lub pramlintidem. Leczenie myszy TgCRND8 AD za pomocą AC253, antagonisty AMYR, lub jego cyklicznego odpowiednika cAC253 zmniejsza neurozapalenie, rozpuszczalne poziomy Aβ i obciążenie blaszek miażdżycowych, poprawiając jednocześnie zdolności poznawcze50. Podobnie, badania in vitro/ex-vivo pokazują, że niskie dawki ludzkiej amyliny lub Aβ powodują zaburzenia LTP i że deficyty te są blokowane przez AC253 lub pramlintid38,39, a wyższe dawki ludzkiej amyliny/ oligomerów amyliny są związane z niekontrolowanym napływem Ca2+, co jest silnie związane ze śmiercią komórek26,32. Łącznie dane te wspierają toksyczną funkcję oligomerów amyliny, a tym samym potencjalny mechanizm terapeutyczny dla blokady AMYR. W przeciwieństwie do tego, inni wykazali, że korzystne efekty amyliny mogą być blokowane za pomocą AC25341. Tak więc, potencjał terapeutyczny leczenia lub hamowania amyliny pozostaje niejasny i podkreśla złożoną i dychotomiczną naturę amyloidów w mózgu i peryferiach.
Piecing Together the Puzzle
W obecnej literaturze istnieje szereg dziur, które wymagają wypełnienia, aby uzyskać pełniejszy obraz historii amyliny: 1) charakter wrodzonego systemu amylinowego i sygnalizacji amylinowej w mózgu 2) możliwości sygnalizacji Aβ i pramlintidu przez trzy główne receptory AMYR i pokrewne 3) mechanizmy terapeutyczne, w których pośredniczy amylina/pramlintid lub hamowanie AMYR. Po pierwsze, interesujące nowe dane wskazują, że AMYR jest zaangażowany nie tylko w sygnalizację, ale także w transport ligandu przez BBB. AMYR jest heterodimerycznym receptorem, który składa się z receptora kalcytoniny i białka modyfikującego aktywność receptora (1-3)51. W tym celu, 50% globalny knockdown receptora kalcytoniny (kluczowego składnika AMYR) znacząco zmniejszył ilość AC253 znalezionego w mózgu50, wskazując, że AMYR zlokalizowane w BBB są zaangażowane w transport tych ligandów do mózgu i mogą być również zaangażowane w przemieszczanie amyliny i pramlintidu do/z mózgu. Istnienie tych mechanizmów transportowych w BBB sugeruje, że amylina prawdopodobnie pełni wrodzoną funkcję fizjologiczną w mózgu, ponieważ jej transport do mózgu jest ściśle kontrolowany. Jednak to, w jaki sposób sygnalizacja amylinowa lub jej brak prowadzi do patologicznych cech AD i czy AMYR jest nośnikiem, poprzez który Aβ pośredniczy w swoim toksycznym działaniu, nadal pozostaje niejasne.
Następnie, istnieją sprzeczne dowody w odniesieniu do związku między Aβ i AMYR. Chociaż kilka badań wyraźnie pokazuje, że ludzka amylina i Aβ mają podobny wpływ na LTP w OUN, a stosowanie inhibitorów AMYR łagodzi te szkodliwe skutki36-39, inne dowody sugerują, że Aβ (1-42) nie jest zdolny do sygnalizacji poprzez AMYR w celu wywołania jakiejkolwiek odpowiedzi cAMP w szerokim zakresie stężeń52. Możliwe jest, że Aβ aktywuje różne kaskady sygnalizacyjne poprzez interakcję z AMYR lub po prostu działa jako obojętny inhibitor kompetycyjny, ale nie zostało to jeszcze wykazane.
Odrębne badanie wykazało ponadto, że oligomeryczna amylina pośredniczy w swoim działaniu toksycznym bezpośrednio przez AMYR i pośrednio przez TRPV4, nieselektywny kanał kationowy26. Niskie stężenia ludzkiej amyliny wywołują odpowiedź na Ca2+, która jest pośredniczona przez jej natywny receptor. Jednak w wyższych stężeniach ludzka amylina tworzy oligomery i uruchamia nieprawidłową sygnalizację, która skutkuje aktywacją kanałów TRVP4 i umożliwia niekontrolowany napływ kationów, zwłaszcza Ca2+. Farmakologiczna blokada AMYR i TRPV4 wykazała, że oba receptory są niezbędne, aby oligomeryczna ludzka amylina mogła wywoływać toksyczne efekty Ca2+26. Jest więc prawdopodobne, że Aβ pośredniczy w swoim toksycznym działaniu na AMYR w podobny sposób, choć takie dane jeszcze nie istnieją. Niekontrolowany napływ Ca2+ jest związany z wieloma zjawiskami patologicznymi, w tym z niekontrolowanym uwalnianiem pęcherzykowym, dysfunkcją OS i mitochondriów, apoptozą itd. W związku z tym jest prawdopodobne, że dysfunkcja komórkowa i rozwój dodatkowych patologii podobnych do AD, które powstają w wyniku sygnalizacji toksycznego amyloidu, są mediowane zarówno przez AMYR, jak i TRPV4. W związku z tym konieczne jest poznanie kaskad sygnałowych, które modulują związek między AMYR i TRVP4. Ponadto, uzasadnione są eksperymenty farmakologiczne, które wykorzystują Aβ i pramlintid w szerokim zakresie dawek, aby określić wpływ Aβ i pramlintidu na prądy Ca2+, LTP, produkcję cAMP i inne kaskady sygnalizacyjne w celu określenia ich zdolności sygnalizacyjnych. Te eksperymenty pomogą wypełnić niektóre z pustych miejsc w obecnej literaturze w odniesieniu do AMYR i jego zaangażowania w stany chorobowe (rysunek 1).
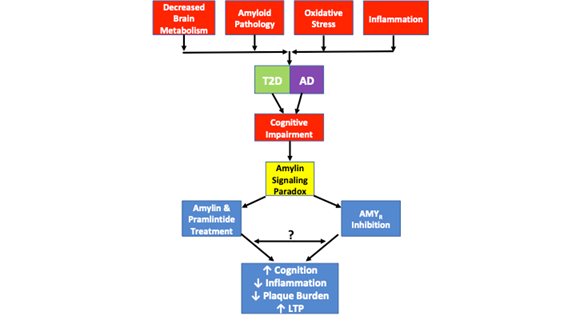
Rysunek 1. przedstawia paradoks sygnalizacji amylinowej i podobieństwa patologiczne obserwowane w T2D i AD. Zmniejszony metabolizm mózgu, patologia amyloidu, stres oksydacyjny i stan zapalny są wspólnymi cechami patologicznymi obserwowanymi w obu chorobach. Chociaż nie każdy przypadek T2D lub AD zawiera każdą z tych cech patologicznych, każdy przypadek wykazuje upośledzenie funkcji poznawczych. Paradoks sygnalizacji amylinowej wchodzi w grę, ponieważ badania wykazały, że zarówno inhibicja AMYR jak i agonizm AMYR poprzez leczenie amyliną i pramlintidem skutkuje poprawą zdolności poznawczych, zmniejszeniem stanu zapalnego, zmniejszeniem obciążenia blaszek miażdżycowych i zwiększeniem LTP. Mechanizmy sygnalizacyjne regulowane przez agonizm i antagonizm AMYR nie zostały jeszcze w pełni wyjaśnione. Podczas gdy sygnalizacja amylinowa jest tradycyjnie kojarzona z sygnalizacją cAMP i PKA, nie jest jasne, czy inne kaskady są również aktywowane przez amylinę/pramlintid. Ponadto nie jest jasne, czy antagoniści AMYR, oligomery amyliny lub Aβ sygnalizują poprzez AMYR lub czy istnieją jakiekolwiek podobieństwa lub wzajemne oddziaływanie pomiędzy wszystkimi tymi ligandami AMYR. W związku z tym szereg eksperymentów zaproponowanych w tym przeglądzie pomoże w dalszym wyjaśnianiu prawdziwej natury AMYR.
Wnioski
Obecne rozbieżności w odniesieniu do roli sygnalizacji amylinowej w mózgu wskazują na istotną potrzebę dalszego wyjaśniania udziału amylin w AD i T2D. W T2D, jest prawdopodobne, że we wczesnych stadiach choroby, amylina zalewa mózg, tworzy oligomery, indukuje nieprawidłową sygnalizację poprzez swój natywny receptor i rekrutuje TRPV4 do indukowania patologicznego napływu Ca2+, co skutkuje rozległą dysfunkcją neuronów, która objawia się jako OS, niekontrolowane uwalnianie pęcherzyków i dysfunkcja interneuronalna, zapalenie, a w konsekwencji śmierć komórek. Mechanizm ten może być odpowiedzialny za początkowe przejście od zdrowego mózgu do starzenia się mózgu w chorobie metabolicznej. W związku z tym, hamowanie AMYR lub TRVP4 w pewnych momentach choroby metabolicznej i we wczesnych stadiach cukrzycy może być uzasadnione w celu zablokowania toksycznego działania oligomerycznej amyliny lub Aβ. Jednakże, silne dowody sugerują również, że zastąpienie amyliny ludzką amyliną lub pramlintidem zmniejsza większość głównych patologii związanych z AD, poprawiając jednocześnie zdolności poznawcze w modelach gryzoni z AD. W związku z tym zastąpienie sygnalizacji amylinowej amyliną lub pramlintydem w środkowych i późnych stadiach cukrzycy, kiedy sygnalizacja amylinowa jest utracona z powodu agregacji, oligomeryzacji lub utraty komórek β, może być uzasadnione. W tym celu istnieje również potrzeba rozeznania czasowej prezentacji zdarzeń patologicznych w metabolicznie powiązanym starzeniu się mózgu oraz opcji terapeutycznych dla wczesnych, pośrednich i późnych stadiów choroby. Krytyczna analiza i badanie bezpośredniej natury i zdolności sygnalizacyjnych tych amyloidów, jak również terapeutycznej natury specyficznych czasowych terapii może pomóc w wypełnieniu luki między terapiami inhibicji AMYR i terapiami zastępującymi amylinę.
Funding
Funding for this article was provided by the National Institutes of Aging grant 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau i dysfunkcja synaptyczna. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013; 80: 1778-1783.
- Association As. 2016 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glucose levels and risk of dementia. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of dementia prevalence in the United States and China. Obesity. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Obesity and its comorbid conditions. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF diabetes atlas. Bruksela: International Diabetes Federation. 2013.
- K Dash S. Cognitive impairment and diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. The lancet Diabetes & endocrinology. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalence of diabetes and prediabetes and their risk factors among Bangladeshi adults: a nationwide survey. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer’s disease. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neuropharmacology. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. The American journal of pathology. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimer’s β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. The American journal of pathology. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylin receptor: a common pathophysiological target in Alzheimer’s disease and diabetes mellitus. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide Antagonizes Beta Amyloid (Aβ)-and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloid-induced depression of hippocampal long-term potentiation is mediated through the amylin receptor. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer’s disease. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylin receptor ligands reduce the pathological cascade of Alzheimer’s disease. Neuropharmacology. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetate injection for the treatment of type 1 and type 2 diabetes mellitus. Clin Ther. 2007; 29: 535-562.
- Singh?Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta?analysis. Diabetes, Obesity and Metabolism. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyclic AC253, a novel amylin receptor antagonist, improves cognitive deficits in a mouse model of Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Activity of pramlintide, rat and human amylin but not Aβ1-42 at human amylin receptors. Endocrinology. 2014; 155: 21-26.