Alexei Drummond, Andrew Rambaut, Remco Bouckaert, and Walter Xie
1 Wstęp
Tutorial ten wprowadza oprogramowanie BEAST do bayesowskiej analizy ewolucyjnej poprzez prosty samouczek. Samouczek obejmuje współestymację filogenezy genów i związanych z nimi czasów dywergencji w obecności informacji kalibracyjnych pochodzących z dowodów kopalnych.
Będziesz potrzebował następującego oprogramowania:
- BEAST – ten pakiet zawiera program BEAST, BEAUti, TreeAnnotator i inne programy użytkowe. Ten tutorial jest napisany dla programu BEAST v2.2.x, który posiada wsparcie dla wielu partycji. Jest on dostępny do pobrania z http://www.beast2.org/.
- Tracer – ten program jest używany do badania wyników programu BEAST (i innych bayesowskich programów MCMC). Graficznie i ilościowo podsumowuje on rozkłady parametrów ciągłych i dostarcza informacji diagnostycznych. W chwili pisania tego tekstu aktualna wersja to v1.6. Można ją pobrać ze strony
http://tree.bio.ed.ac.uk/software/. - FigTree – jest to aplikacja do wyświetlania i drukowania filogenez molekularnych, w szczególności tych uzyskanych za pomocą programuBEAST. W momencie pisania tego tekstu, aktualna wersja to v1.4.2. Jest ona dostępna do pobrania z http://tree.bio.ed.ac.uk/software/.
Tutorial ten poprowadzi Cię przez analizę wyrównania sekwencji pobranych z dwunastu gatunków naczelnych (patrz Rysunek 1). Celem jest oszacowanie filogenezy, tempa ewolucji każdej linii i wieku nieskalibrowanych różnic międzygatunkowych.
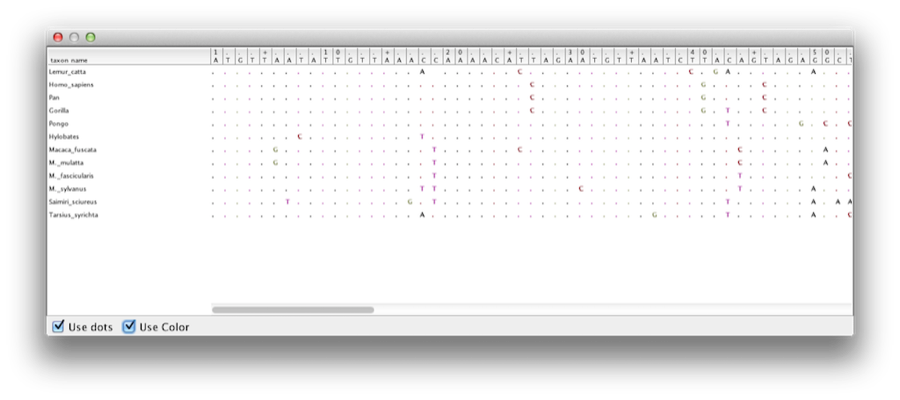
Rysunek 1: Część wyrównania dla naczelnych.
Pierwszym krokiem będzie konwersja pliku NEXUS z blokiem DATA lub CHARACTERS do pliku wejściowego BEAST XML. Można to zrobić za pomocą programu BEAUti (skrót od Bayesian Evolutionary Analysis Utility). Jest to przyjazny dla użytkownika program do ustawiania modelu ewolucyjnego i opcji dla analizy MCMC. Drugim krokiem jest faktyczne uruchomienie BEAST przy użyciu pliku wejściowego wygenerowanego przez BEAUTi, który zawiera dane, model i ustawienia analizy. Ostatnim krokiem jest zbadanie danych wyjściowych BEAST w celu zdiagnozowania problemów i podsumowania wyników.
2 BEAUti
Program BEAUti jest przyjaznym dla użytkownika programem do ustawiania parametrów modelu dla BEAST. Uruchom BEAUti klikając dwukrotnie na jego ikonę. Po uruchomieniu, BEAUti będzie wyglądał podobnie niezależnie od systemu komputerowego, na którym jest uruchomiony. W tym poradniku na rysunkach użyto wersji Mac OS X, ale wersje dla Linux i Windows będą miały ten sam układ i funkcjonalność.
2.1 Wczytanie pliku NEXUS
Aby wczytać wyrównanie w formacie NEXUS, wystarczy wybrać opcję Importuj wyrównanie… z menu Plik, lub przeciągnąć plik na środek panelu Partycje.
Przykładowy plik o nazwie primate-mtDNA.nex jest dostępny w katalogu examples/nexus/ dla Mac i Linux oraz examples/nexus/ dla Windows wewnątrz katalogu, w którym został zainstalowany BEAST.Plik ten zawiera wyrównanie sekwencji 12 gatunków naczelnych.
Okno Add Partition (Rysunek 2) pojawi się, jeśli powiązany pakiet jest zainstalowany. Jeśli używasz „czystego” BEAST 2, możesz przejść do następnego akapitu. W przeciwnym razie, wybierz Dodaj wyrównanie i kliknij OK, aby kontynuować.
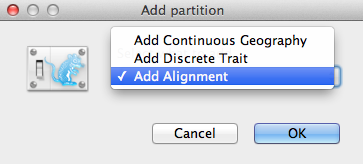
Rysunek 2: Okno Dodaj partycję (pojawia się tylko wtedy, gdy zainstalowane są powiązane pakiety).
Jeśli istnieje jakiekolwiek nakładanie się kodowania w partycjach, pojawi się okno komunikatu ostrzegawczego (Rysunek 3). Należy je przeczytać i kliknąć OK, aby kontynuować.
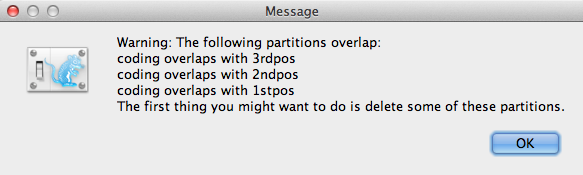
Rysunek 3: Okno komunikatu ostrzegawczego (pojawia się tylko wtedy, gdy w partycjach występuje nakładanie się kodowania).
Po załadowaniu, w panelu głównym wyświetlane są pięcioznakowe partycje (Rysunek 4). Wyrównanie jest podzielone na część kodującą białko i część niekodującą, a część kodująca jest podzielona na pozycje kodonów 1, 2 i 3. Przed przejściem do następnego kroku należy usunąć partycję 'coding’, ponieważ odnosi się ona do tych samych nukleotydów co partycje '1stpos’, '2ndpos’ i '3rdpos’. Aby usunąć partycję „coding”, należy wybrać wiersz i kliknąć przycisk „-” na dole tabeli. Wyrównanie można obejrzeć klikając dwukrotnie na partycję.
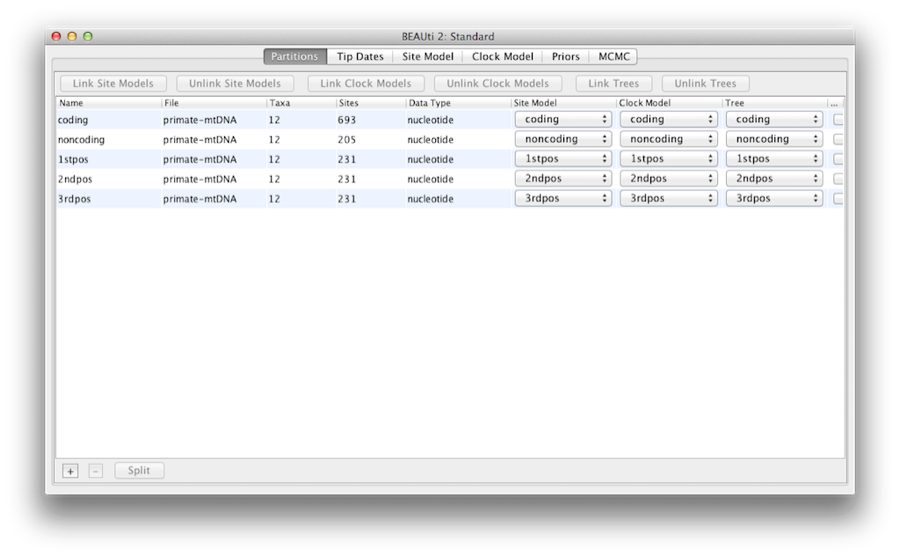
Rysunek 4: Zrzut ekranu zakładki dane w BEAUti. Ten i wszystkie następne zrzuty ekranu zostały wykonane na komputerze Apple z systemem Mac OS X i będą wyglądać nieco inaczej w innych systemach operacyjnych.
Łączenie/odłączanie modeli partycji
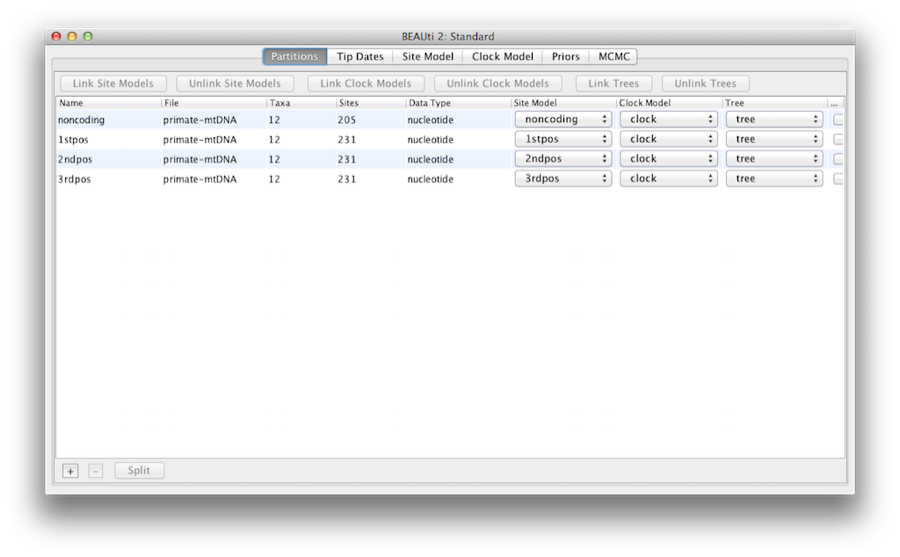
Rysunek 5: Zrzut ekranu zakładki Partycje w BEAUti po połączeniu i zmianie nazwy modelu zegara i drzewa.
Ponieważ sekwencje są połączone (tzn. wszystkie pochodzą z genomu mitochondrialnego, który, jak się uważa, nie ulega rekombinacji u ptaków i ssaków), mają tego samego przodka, więc partycje powinny mieć to samo drzewo czasowe w modelu. Dla uproszczenia, założymy również, że partycje mają to samo tempo ewolucji dla każdej gałęzi, a więc ten sam „model zegara”. Ograniczymy nasze modelowanie heterogeniczności tempa do heterogeniczności pomiędzy miejscami w obrębie każdej partycji, a także pozwolimy, aby partycje miały różne średnie tempo ewolucji.
W tym momencie musimy więc połączyć model zegara i drzewo. W panelu Partitions zaznaczamy wszystkie cztery partycje w tabeli (lub żadną, domyślnie wszystkie partycje są dotknięte) i klikamy przycisk Link Trees, a następnie Link Clock Models (patrz Rysunek 5). Następnie kliknij pierwsze menu rozwijane w kolumnie Clock Model i zmień nazwę współdzielonego modelu zegara na 'clock’. Podobnie zmień nazwę współdzielonego drzewa na 'tree’. Dzięki temu kolejne opcje i wygenerowane pliki dziennika będą łatwiejsze do odczytania.
2.2 Ustawianie modelu zastępczego
Kolejnym krokiem jest ustawienie modelu zastępczego. Następnie należy wybrać zakładkę Site Models na górze głównego okna (pomijamy zakładkę Tip Dates, ponieważ wszystkie taksony pochodzą z próbek współczesnych). Spowoduje to wyświetlenie ustawień modelu ewolucyjnego dla BEAST. Dostępne opcje zależą od tego, czy dane są nukleotydami, czy aminokwasami, danymi binarnymi, czy danymi ogólnymi. Ustawienia, które pojawią się po załadowaniu wyrównania nukleotydów naczelnych będą wartościami domyślnymi dla danych nukleotydowych, więc musimy dokonać pewnych zmian.
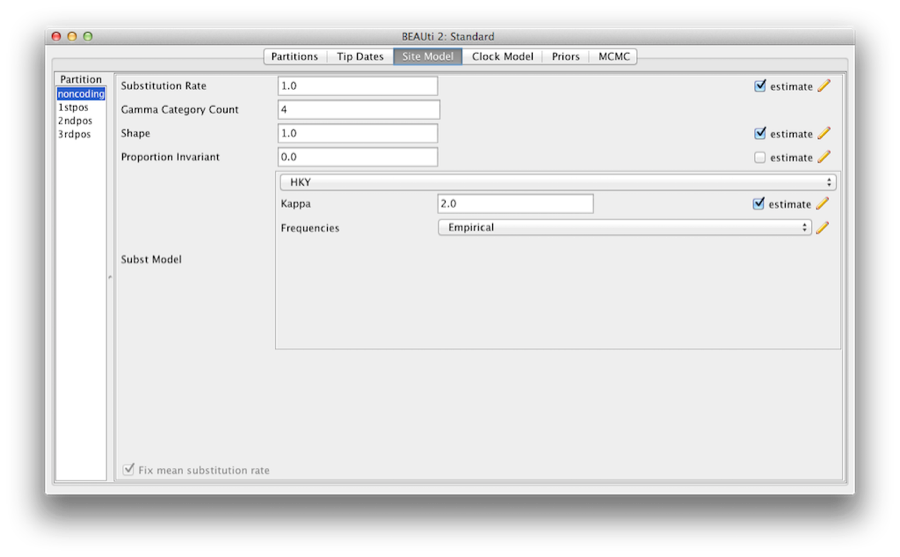
Rysunek 6: Zrzut ekranu zakładki site model w BEAUti.
Większość modeli powinna być Państwu znana. Najpierw należy ustawić Gamma Category Count na 4, a następnie zaznaczyć pole „estymacja” dla parametru Shape. Pozwoli to na modelowanie zmienności współczynnika pomiędzy miejscami w każdej partycji. Zauważ, że 4 do 6 kategorii działa wystarczająco dobrze dla większości zbiorów danych, podczas gdy posiadanie większej liczby kategorii wymaga więcej czasu na obliczenia dla niewielkiej dodatkowej korzyści. Pozostawiamy pozycję Proportion Invariant ustawioną na zero.
Następnie wybieramy HKY z menu rozwijanego Subst Model. Idealnie byłoby wybrać model substytucji, który najlepiej pasuje do danych dla każdej partycji, ale tutaj dla uproszczenia używamy HKY dla wszystkich partycji. Następnie wybierz Empirical z menu rozwijanego Frequencies. Spowoduje to dopasowanie częstości do proporcji zaobserwowanych w danych (dla każdej partycji osobno, gdy już rozłączymy modele miejsc). Takie podejście oznacza, że możemy uzyskać dobre dopasowanie do danych bez wyraźnego szacowania tych parametrów. Robimy to tutaj tylko po to, by pliki dziennika były nieco krótsze i bardziej czytelne w dalszej części ćwiczenia.
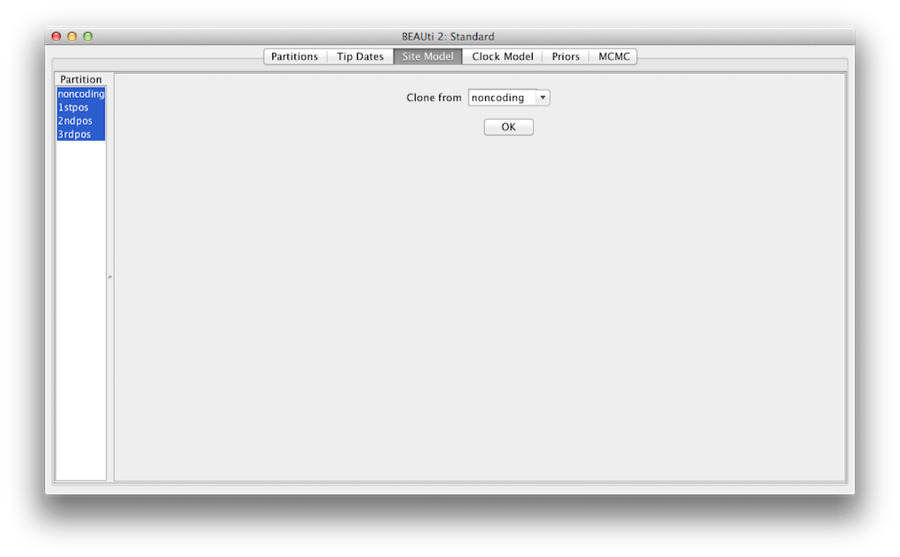
Rysunek 7: konfiguracja klonów z jednego modelu strony do innych.
Na koniec zaznaczamy pole 'estymuj’ dla parametru szybkości substytucji i zaznaczamy pole wyboru Ustal średnią szybkość mutacji. Dzięki temu poszczególne partycje będą miały oszacowane względne tempo dla niepołączonych modeli witryn (Rysunek 6).
Na koniec przytrzymaj klawisz 'shift’, aby wybrać wszystkie modele witryn po lewej stronie i kliknij OK, aby sklonować ustawienia z niekodowania do 1stpos, 2ndpos i 3rdpos (Rysunek 7). Przejdź przez każdy model strony, jak widać, ich konfiguracje są teraz takie same.
2.3 Ustawianie modelu zegara
Kolejnym krokiem jest wybranie zakładki Clock Models w górnej części okna głównego. W tym miejscu wybieramy model zegara molekularnego. W tym ćwiczeniu pozostawimy domyślną wartość ścisłego zegara molekularnego, ponieważ dane te są bardzo podobne do zegara i nie wymagają zmienności tempa między gałęziami, aby mogły być uwzględnione w modelu.
Aby przetestować podobieństwo do zegara, można (i) przeprowadzić analizę przy użyciu zrelaksowanego modelu zegara i sprawdzić, jak bardzo zmienność stawek jest implikowana przez dane (zobacz współczynnik zmienności, aby dowiedzieć się więcej na ten temat), lub (ii) przeprowadzić porównanie modelu między ścisłym i zrelaksowanym zegarem przy użyciu pathsampling, lub (iii) użyć modelu losowego zegara lokalnego, który wyraźnie rozważa, czy każda gałąź w drzewie potrzebuje własnego wskaźnika gałęzi.
2.4 Priorytety
Karta Priorytety pozwala na określenie priorytetów dla każdego parametru w modelu. Wybór modelu w zakładkach Model miejsca i Model zegara skutkuje włączeniem różnych parametrów do modelu, które są widoczne w zakładce Priors (patrz Rysunek 8).
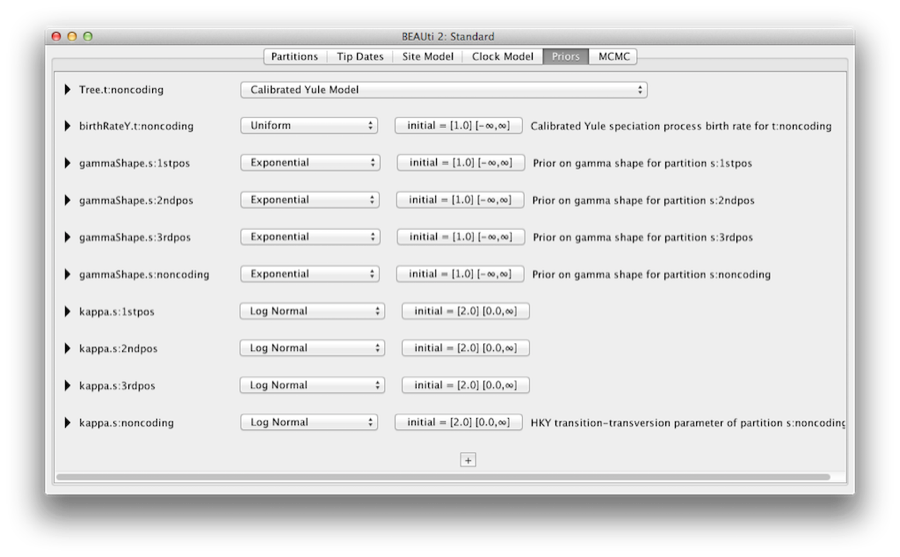
Rysunek 8: Zrzut ekranu zakładki Priors w BEAUti.
W tym miejscu określamy również, że chcemy użyć skalibrowanego modelu Yule jako priorytetu drzewa. Model Yule’a jest prostym modelem specjacji, który jest generalnie bardziej odpowiedni, gdy rozważamy sekwencje pochodzące od różnych gatunków. Wybierz Calibrated Yule Model z rozwijanego menu Tree prior.
2.4.1 Definiowanie węzła kalibracji
Musimy teraz określić rozkład priorytetu na kalibrowanym węźle, w oparciu o naszą wiedzę o skamielinach. Jest to znane jako kalibracja naszego drzewa. Aby zdefiniować dodatkowy priorytet, naciśnij mały przycisk + poniżej listy priorytetów. Jeśli nie jest on widoczny w Twoim widoku, przewiń panel do dołu, aby znaleźć przycisk +. Zobaczysz okno dialogowe, które pozwoli Ci zdefiniować podzbiór taksonów w drzewie filogenetycznym. Po utworzeniu zestawu taksonów będziesz mógł później dodać informacje kalibracyjne dla jego najnowszego wspólnego przodka (MRCA).
Nadaj nazwę zbiorowi taksonów, wypełniając pole etykiety zbioru taksonów. Nazwij go human-chimp, ponieważ będzie on zawierał taksony dla Homo sapiens i Pan. Na poniższej liście zobaczysz dostępne taksony. Zaznacz po kolei każdy z dwóch taksonów i naciśnij przycisk strzałki > >. (Rysunek 9).Kliknij przycisk OK, a nowo zdefiniowany zestaw taksonów zostanie dodany do listy predyktorów.Ponieważ jest to skalibrowany węzeł, który ma być używany w połączeniu ze skalibrowanym predyktorem Yule, należy wymusić monofiletyczność, więc zaznacz pole wyboru oznaczone jako Monophyletic. To ograniczy topologię drzewa w taki sposób, że grupa człowiek-szympans będzie monofiletyczna w trakcie analizy MCMC.
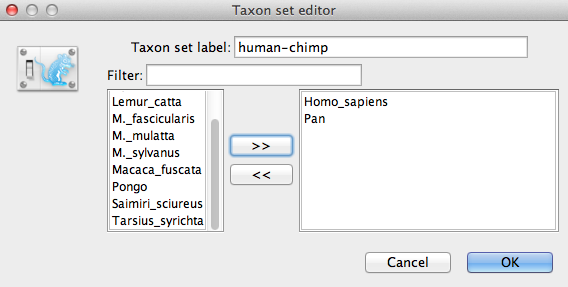
Rysunek 9: Edytor zestawu taksonów w BEAUti.
Aby zakodować informacje o kalibracji, musimy określić rozkład dla MRCA człowieka-szympansa.Wybierz rozkład logarytmiczno-normalny z rozwijanego menu po prawej stronie nowo dodanego human-chimp.prior. Kliknij na czarny trójkąt, a pojawi się wykres funkcji gęstości prawdopodobieństwa wraz z parametrami rozkładu logarytmiczno-normalnego. Ustawimy M=1,78 i S=0,085, co da nam rozkład wyśrodkowany na około 6 mln lat z odchyleniem standardowym około 0,5 mln lat. Da to centralny 95% zakres prawdopodobieństwa obejmujący 5-7 Mya. Odpowiada to z grubsza aktualnemu szacunkowi daty ostatniego wspólnego przodka człowieka i szympansa (Rysunek 10).
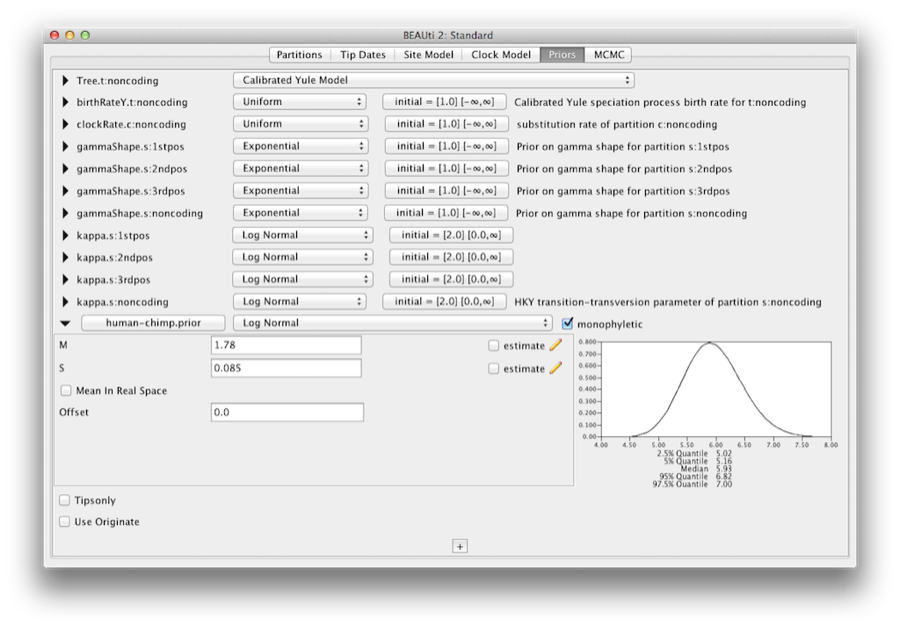
Rysunek 10: Zrzut ekranu opcji priorytetu kalibracji w panelu Priors w BEAUti.
Powinniśmy się przekonać, że priorytety pokazane w panelu priorytety rzeczywiście odzwierciedlają informacje o parametrach modelu, które posiadamy. W końcu określimy również pewne rozproszone, „nieinformacyjne”, ale właściwe priorytety na ogólną szybkość zegara molekularnego (clockRate) i szybkość specjacji (birthRateY) priorytetu drzewa Yule. Dla każdego z nich wybieramy z rozwijanego menu opcję Gamma i za pomocą przycisku strzałki rozwijamy widok, aby ujawnić parametry priorytetu Gamma. Zarówno dla współczynnika zegarowego, jak i współczynnika urodzeń Yule ustaw parametr Alpha(shape) na 0,001, a parametr Beta (scale) na 1000 (rysunek 11).
Domyślnie każdy z parametrów kształtu gamma ma wykładniczy rozkład priordystrybucji ze średnią 1. Oznacza to (patrz rysunek 3.7), że spodziewamy się nieco mniejszej zmienności. Domyślnie parametry kappa dla modelu HKY mają rozkład wstępny lognormalny(1,1.25), co zasadniczo zgadza się z empirycznymi dowodami dotyczącymi zakresu realistycznych wartości dla skośności przejścia/przejścia. Zachowano te domyślne priorytety, ponieważ są one odpowiednie dla tej konkretnej analizy.
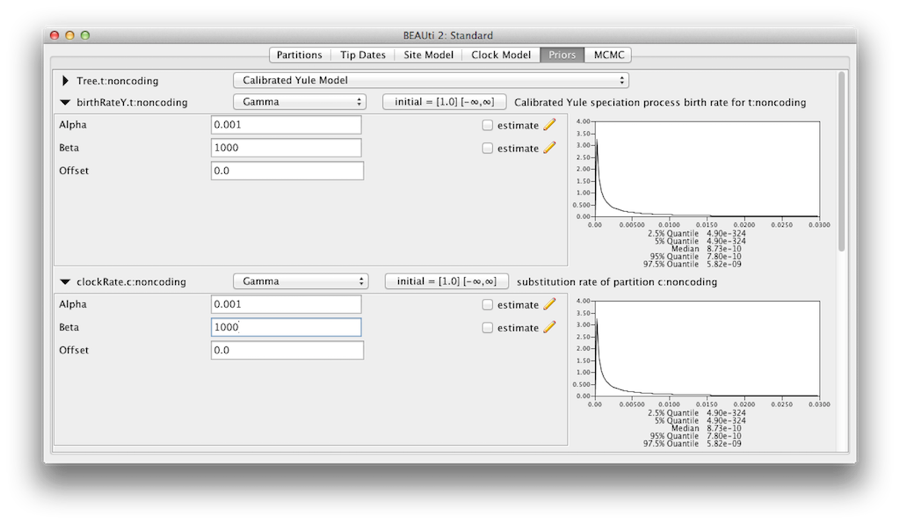
Rysunek 11: Priorytet gamma.
2.5 Ustawianie opcji MCMC
Kolejna zakładka, MCMC, zawiera bardziej ogólne ustawienia pozwalające kontrolować długość przebiegu MCMC i nazwy plików.
Po pierwsze mamy Długość łańcucha. Jest to liczba kroków, które MCMC wykona w łańcuchu przed jego zakończeniem. Długość łańcucha zależy od wielkości zbioru danych, złożoności modelu i wymaganej jakości odpowiedzi. Domyślna wartość 10 000 000 jest całkowicie arbitralna i powinna być dostosowana do wielkości zbioru danych. Dla tego zestawu danych ustawmy długość łańcucha na 6 000 000, ponieważ będzie to działało dość szybko na większości nowoczesnych komputerów (kilka minut).
Pole Store Every określa jak często stan jest zapisywany do pliku. Okresowe zapisywanie stanu jest przydatne w sytuacjach, gdy środowisko obliczeniowe nie jest zbyt niezawodne i działanie BEAST może zostać przerwane. Posiadanie zapisanej kopii ostatniego stanu pozwala na wznowienie łańcucha zamiast rozpoczynania go od początku, tak więc nie ma potrzeby ponownego przechodzenia przez burn-in. Pole Pre Burnin określa liczbę próbek, które nie są rejestrowane na samym początku analizy. Pola Store Every i Pre Burnin pozostawiamy ustawione na wartości domyślne. Poniżej znajdują się szczegóły plików logów. Każdą z nich można rozwinąć klikając na czarny trójkąt.
Kolejne opcje określają jak często wartości parametrów w łańcuchu Markowa powinny być wyświetlane na ekranie i zapisywane w pliku dziennika.Wyjście ekranowe służy tylko do monitorowania postępu programu i może być ustawione na dowolną wartość (chociaż jeśli jest ustawione na zbyt małą, sama ilość informacji wyświetlanych na ekranie spowolni program). Dla pliku dziennika, wartość powinna być ustawiona w odniesieniu do całkowitej długości łańcucha. Zbyt częste próbkowanie spowoduje powstanie bardzo dużych plików z niewielką dodatkową korzyścią w postaci dokładności analizy. Zbyt rzadkie próbkowanie spowoduje, że plik dziennika nie będzie zawierał wystarczających informacji o rozkładach parametrów. Prawdopodobnie chcesz dążyć do przechowywania nie więcej niż 10,000 próbek, więc to powinno być ustawione na nie mniej niż długość łańcucha / 10,000.
Dla tego ćwiczenia ustawimy częstotliwość dziennika śladu i dziennika drzewa na 1,000 i dziennika ekranu na 10,000.Określ również Primates.log jako nazwę pliku dziennika śladu i Primates.trees jako nazwę pliku dziennika drzewa.Upewnij się, że nazwa pliku dziennika ekranowego jest pusta, w przeciwnym razie dziennik ekranowy nie będzie zapisywany na ekranie.
- Jeśli używasz systemu operacyjnego Windows, sugerujemy dodanie końcówki .txt do obu tych plików (Primates.log.txt i Primates.trees.txt), aby Windows rozpoznał je jako pliki tekstowe.
2.6 Generowanie pliku BEAST XML
Jesteśmy teraz gotowi do utworzenia pliku BEAST XML. W tym celu wybieramy opcję Zapisz z menu Plik. Zaznaczamy domyślne priorytety i zapisujemy plik z odpowiednią nazwą (zwykle kończymy nazwę pliku na .xml, np. Primates.xml).Teraz jesteśmy gotowi do uruchomienia pliku przez BEAST.
3 Uruchamianie programu BEAST
Rysunek 12: Zrzut ekranu programu BEAST.
Następnie uruchamiamy program BEAST i gdy zostanie on poproszony o podanie pliku wejściowego, podajemy nowo utworzony plik XML jako dane wejściowe. BEAST będzie działał aż do zakończenia raportowania informacji na ekranie. Pliki z rzeczywistymi wynikami zostaną zapisane na dysku w tej samej lokalizacji co plik wejściowy. Wyjście na ekran będzie wyglądało następująco:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Zauważ, że na początku znajduje się kilka użytecznych informacji dotyczących dopasowań oraz tego, jakie prawdopodobieństwa drzew są używane. Również, wszystkie cytaty istotne dla analizy są wymienione na początku przebiegu, co może być łatwo skopiowane do rękopisów raportujących o analizie. Następnie następuje raportowanie łańcucha, co daje pewne informacje zwrotne w czasie rzeczywistym o postępie łańcucha.
Na końcu drukowana jest analiza operatorów, która zawiera listę wszystkich operatorów użytych w analizie wraz z informacją, jak często dany operator był próbowany, akceptowany i odrzucany (patrz kolumny #total, #accept i #reject odpowiednio). Współczynnik akceptacji to proporcja przypadków, w których operator jest akceptowany, gdy jest wybrany do złożenia propozycji. Ogólnie rzecz biorąc, współczynnik akceptacji, który jest wysoki, powiedzmy powyżej 0.5 wskazuje, że propozycje są konserwatywne i nie eksplorują przestrzeni parametrów w sposób efektywny. Z drugiej strony niski współczynnik akceptacji wskazuje, że propozycje są zbyt agresywne i prawie zawsze prowadzą do stanu, który jest odrzucany ze względu na niski posterior.Zarówno zbyt wysoki jak i zbyt niski współczynnik akceptacji skutkuje niskimi wartościami ESS. Współczynnik akceptacji 0,234 jest celem (opartym na bardzo ograniczonych dowodach dostarczonych przez ) dla wielu (ale nie wszystkich) operatorów zaimplementowanych w BEAST.
Niektóre operatory mają parametr dostrajania, na przykład współczynnik skali parametru ascale. Jeśli ostateczny wskaźnik akceptacji nie jest bliski docelowemu, BEAST zasugeruje nową wartość dla parametru dostrajania, która jest drukowana w analizie operatora. W tym przypadku, wszystkie wskaźniki akceptacji są dobre dla operatorów, którzy mają parametry dostrajania. Operatory bez parametrów dostrajających obejmują operatory Wideexchange i Wilson-Balding dla tej analizy. Oba te operatory próbują zmieniać topologię drzewa z dużymi krokami, ale ponieważ dane w przeważającej mierze wspierają pojedynczą topologię, te radykalne propozycje są prawie zawsze odrzucane.
4 Analizowanie wyników
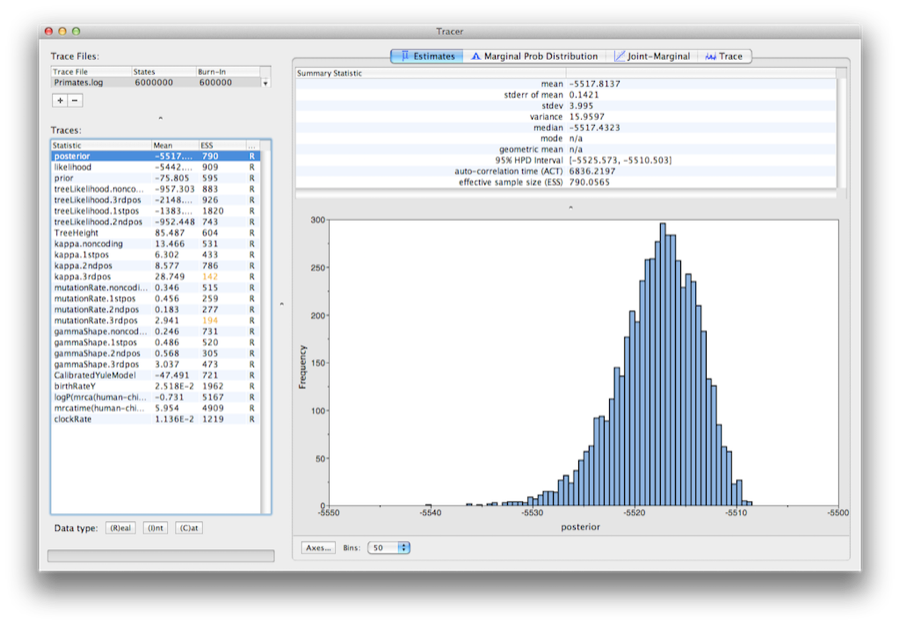
Rysunek 13: Zrzut ekranu programu Tracer v1.6.
Uruchomić program o nazwie Tracer w celu przeanalizowania danych wyjściowych programu BEAST. Po otwarciu głównego okna, wybierz Import Trace File… z menu File i wybierz plik utworzony przez BEAST o nazwie Primates.log (Rysunek 13).
Pamiętaj, że MCMC jest algorytmem stochastycznym, więc rzeczywiste liczby nie będą dokładnie takie same jak te przedstawione na rysunku.
Po lewej stronie znajduje się lista różnych wielkości, które BEAST zapisał do pliku. Są tam ślady dla posterior (jest to logarytm naturalny iloczynu prawdopodobieństwa drzewa i gęstości priorytetu) i parametrów ciągłych. Wybranie śladu po lewej stronie powoduje wyświetlenie analiz dla tego śladu po prawej stronie, w zależności od wybranej karty. Przy pierwszym otwarciu wybierany jest ślad 'posterior’, a różne statystyki tego śladu wyświetlane są w zakładce Estimates.W prawym górnym rogu okna znajduje się tabela z obliczonymi statystykami dla wybranego śladu.
Wybierz parametr clockRate z listy po lewej stronie, aby spojrzeć na średnie tempo ewolucji (uśrednione dla całego drzewa i wszystkich miejsc). Tracer wykreśli histogram (marginal posterior) dla wybranej statystyki, a także poda statystyki podsumowujące, takie jak średnia i mediana. 95% HPD oznacza przedział o największej gęstości następczej i reprezentuje najbardziej zwarty przedział dla wybranego parametru, który zawiera 95% prawdopodobieństwa następczego. Można go traktować jako bayesowski odpowiednik przedziału ufności. Parametr TreeHeight daje marginalny rozkład potomny wieku korzenia całego drzewa.
Zaznacz parametr TreeHeight, a następnie kliknij Ctrl-click mrcatime(human-chimp) (Command-click w Mac OS X). Spowoduje to wyświetlenie wieku korzenia i kalibracji MRCA, którą podaliśmy wcześniej w BEAUti. Można sprawdzić, że dywergencja, której użyliśmy do kalibracji drzewa(mrcatime(human-chimp)) ma rozkład potomny, który pasuje do określonego przez nas rozkładu wcześniejszego (Rysunek 14).
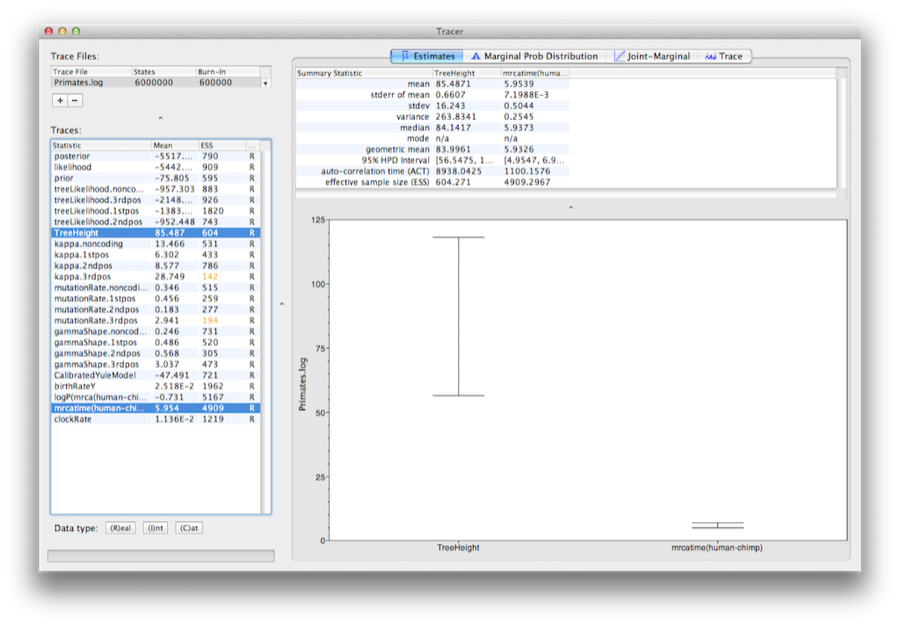
Rysunek 14: Zrzut ekranu 95% przedziałów HPD wysokości korzenia i MRCA określonego przez użytkownika (human-chimp) w programie Tracer.
5 Marginal posterior estimates
Aby pokazać względne współczynniki dla czterech partycji, należy zaznaczyć parametr mutationRate dla każdej z czterech partycji i wybrać zakładkę gęstości marginalnej w Tracer.Rysunek 15 pokazuje gęstości marginalne dla względnych współczynników substytucji. Theplot pokazuje, że pozycje kodonu 1 i 2 mają znacząco różne stawki (0.456versus 0.183) i oba są znacznie wolniejsze niż pozycja kodonu 3 z względną stawką 2.941. Partycja niekodująca ma tempo pośrednie między pozycjami kodonów 1 i 2 (0,346). Łącznie wyniki te sugerują silną selekcję oczyszczającą zarówno w regionach kodujących, jak i niekodujących dopasowania.
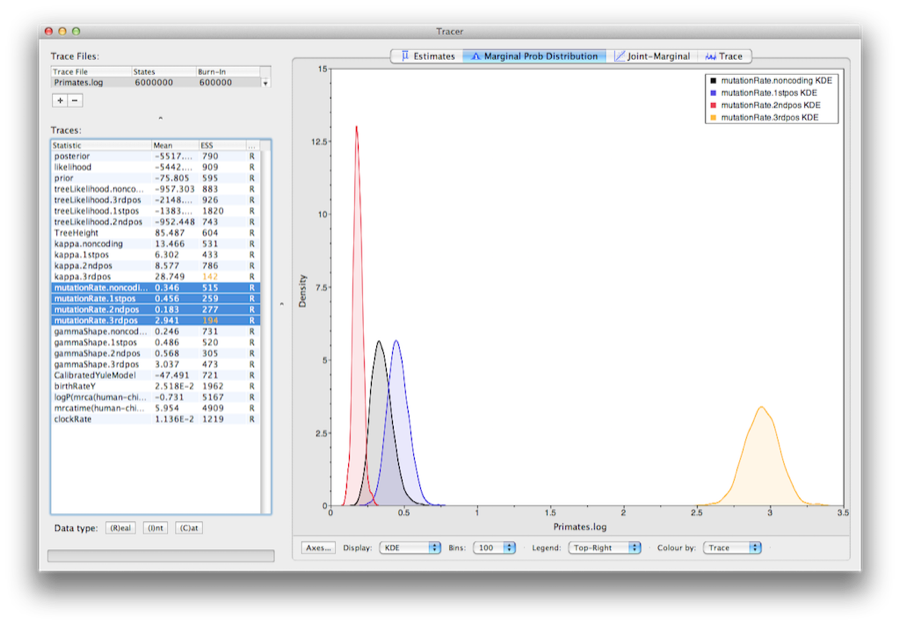
Ryc. 15: Zrzut ekranu z marginalnymi gęstościami potomnymi względnych współczynników substytucji w czterech partycjach (względem średniej ważonej współczynnikiem).
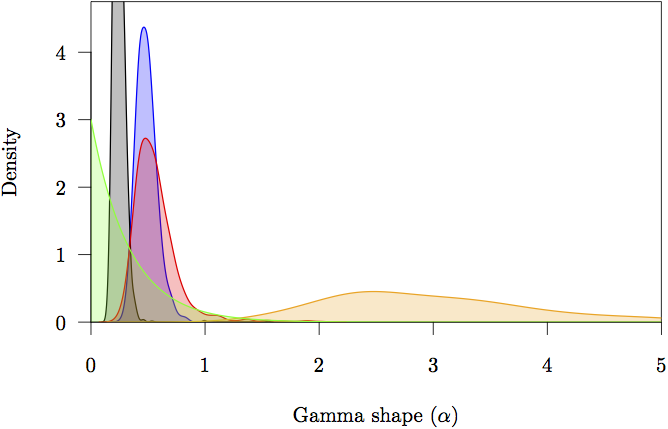
Rysunek 16: Krańcowe gęstości wstępne i potomne dla parametrów kształtu (α). Prior jest zaznaczony kolorem szarym. Pokazano również oszacowanie gęstości potomnej dla każdej partycji: niekodującej (pomarańczowa) oraz pierwszej (czerwona), drugiej (zielona) i trzeciej (niebieska) pozycji kodonu.
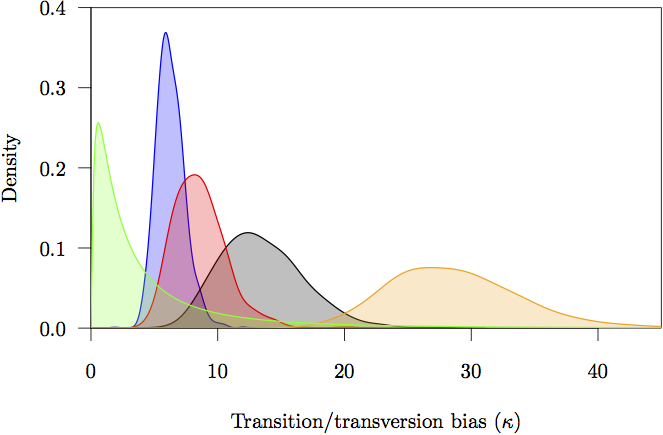
Rysunek 17: Krańcowe wartości priorytetu i gęstości potomnej dla parametrów skośności przejścia/transwersji (κ). Prior jest zaznaczony kolorem szarym. Pokazano również oszacowanie gęstości potomnej dla każdej partycji: niekodującej (pomarańczowa) oraz pozycji pierwszego (czerwona), drugiego (zielona) i trzeciego (niebieska) kodonu.
Pytania
Jakie jest szacowane tempo ewolucji molekularnej dla tego drzewa genów (uwzględnij 95% przedział HPD)?
Jakie źródła błędu zawiera to oszacowanie?
Jak stary jest korzeń drzewa (podać średnią i 95% przedział HPD)?
6 Uzyskanie oszacowania drzewa filogenetycznego
BEAST produkuje również potomną próbkę filogenetycznych drzew czasowych wraz z próbką oszacowań parametrów. Muszą one być podsumowane przy użyciu programu TreeAnnotator. Program ten weźmie zestaw drzew i znajdzie najlepiej wspierane. Następnie doda adnotacje do tego reprezentatywnego drzewa podsumowującego, podając średni wiek wszystkich węzłów i odpowiadające im 95% zakresy HPD. Obliczy również prawdopodobieństwo kladu dla każdego węzła. Uruchom program TreeAnnotator i skonfiguruj go w sposób przedstawiony na Rysunku 18.
Rysunek 18: Zrzut ekranu programu TreeAnnotator.
Curnin to liczba drzew do usunięcia z początku próbki. W przeciwieństwie do Tracera, który określa liczbę kroków jako burnin, w TreeAnnotator musisz określić rzeczywistą liczbę drzew. Dla tego przebiegu określono długość łańcucha 6,000,000 kroków, próbkując co 1,000 kroków. W związku z tym plik z drzewami będzie zawierał 6000 drzew, a więc należy określić 10% burnin w górnym polu tekstowym.
Opcja Posterior probability limit określa limit, że jeśli węzeł występuje z mniejszą częstotliwością niż ta częstotliwość w próbce drzew (tj. ma prawdopodobieństwo posterior mniejsze niż ten limit), nie zostanie opisany. Domyślna wartość 0.5 oznacza, że tylko węzły występujące w większości drzew będą opisywane. Ustaw to na zero, aby opisać wszystkie węzły.
Typ drzewa docelowego określa topologię drzewa, które będzie opisywane. Możesz wybrać konkretne drzewo z pliku lub poprosić TreeAnnotator o znalezienie drzewa w twojej próbce.Domyślna opcja, Maximum clade credibility tree, znajduje drzewo z najwyższym iloczynem prawdopodobieństwa potomnego wszystkich jego węzłów.
Dla wysokości węzłów, domyślną opcją jest Common Ancestor Heights, która oblicza wysokość węzła jako średnią czasu MRCA wszystkich par węzłów w kladzie. W przypadku drzew o dużej niepewności topologicznej, a więc wielu kladów o niskim wsparciu, niektóre inne metody mogą prowadzić do uzyskania drzew o ujemnych długościach gałęzi. W tej analizie wsparcie dla wszystkich kladów w drzewie sumarycznym jest bardzo wysokie, więc nie stanowi to problemu.Wybierz Średnie wysokości dla wysokości węzłów. To ustawia wysokość (wiek) każdego węzła w drzewie na średnią wysokość w całej próbie drzew dla tego kladu.
Dla pliku wejściowego wybierz plik drzew utworzony przez BEAST i wybierz plik wyjściowy (tutaj nazwaliśmy go Primates.MCC.tree). Teraz naciśnij Run i poczekaj aż program zakończy pracę.
7 Wizualizacja oszacowania drzewa
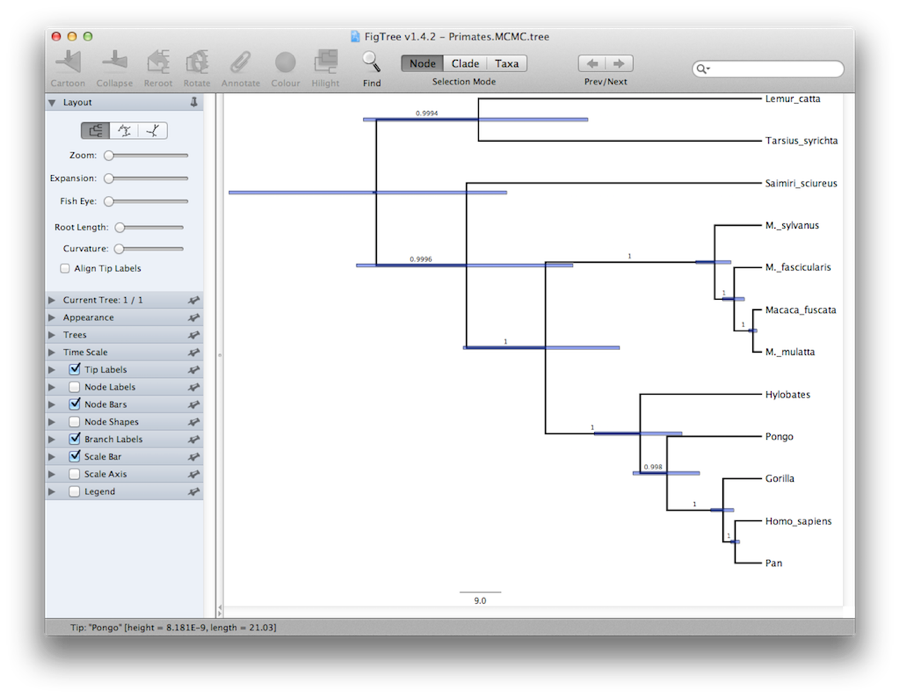
Na koniec, możemy zwizualizować drzewo w innym programie o nazwie FigTree. Uruchom ten program i otwórz plik Primates.MCC.tree za pomocą polecenia Open w menu File. Teraz możesz spróbować wybrać kilka opcji w panelu kontrolnym po lewej stronie. Po pierwsze, odrzuć opcję Trees w panelu, zaznacz Order nodes i wybierz Ordering by decreasing. Spróbuj wybrać opcję Node Bars, aby uzyskać paski błędów wieku węzłów. Włącz również opcję Branch Labels i wybierz posterior, aby uzyskać wyświetlanie prawdopodobieństwa potomnego dla każdego węzła. Jeśli używasz modelu zegara, który nie jest ścisły, to w sekcji Wygląd możesz również powiedzieć FigTree, aby pokolorowało gałęzie według tempa. Powinieneś skończyć z czymś podobnym do Rysunku 19.
Rysunek 19: Zrzut ekranu FigTree i DensiTree.
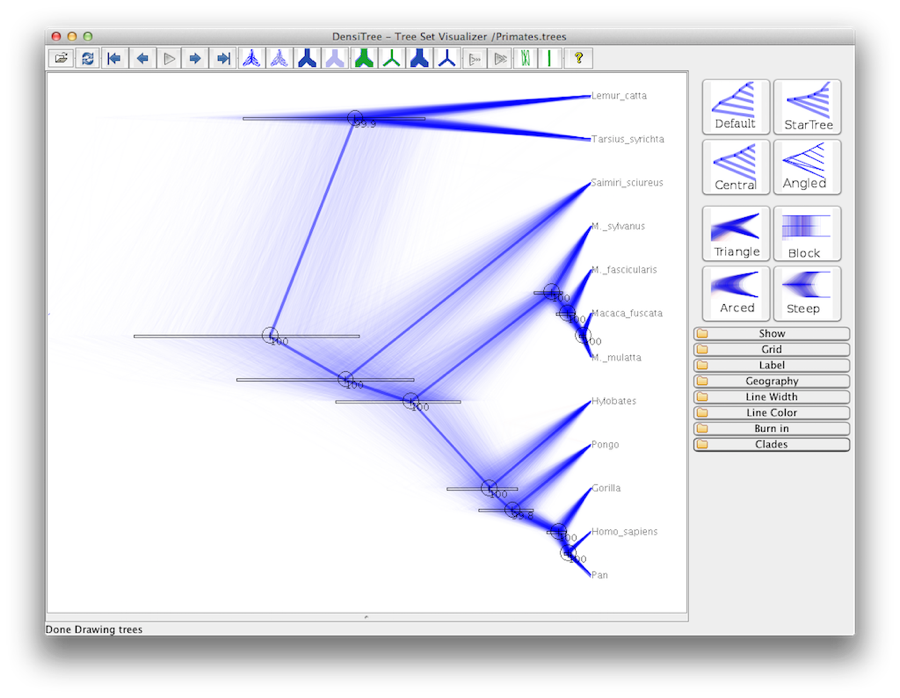
Alternatywny widok drzewa może być wykonany za pomocą DensiTree, który jest częścią Beast 2. Zaletą DensiTree jest to, że jest w stanie wizualizować zarówno niepewność w wysokości węzłów jak i niepewność w topologii.Dla tego konkretnego zestawu danych, dominująca topologia jest obecna w ponad 99% próbek. Tak więc wnioskujemy, że ta analiza skutkuje bardzo wysokim konsensusem co do topologii (Rysunek 19).
Pytania
- Czy tempo ewolucji różni się znacząco wśród różnych linii w drzewie?
- DensiTree ma pasek kladów (Menu Window/View clade toolbar), aby pokazać informacje o kladach.
Jakie jest wsparcie dla kladu ?
- Możesz przeglądać topologie w DensiTree używając menu Browse.Najbardziej popularna topologia ma wsparcie ponad 99%.
Jakie jest wsparcie dla drugiej najbardziej popularnej topologii?
- Pod menu Pomoc, DensiTree pokazuje pewne informacje.
Ile topologii jest w zestawie drzew?
8 Porównanie wyników do priorytetu
Dobrym pomysłem jest ponowne przeprowadzenie analizy przy próbkowaniu z priorytetu, aby upewnić się, że interakcje między priorytetami nie mają wpływu na informacje o priorytecie. Interakcja między priorytetami może być problematyczna, szczególnie gdy używamy kalibracji, ponieważ oznacza to nałożenie wielu priorytetów na drzewo.
Używając BEAUti, skonfiguruj tę samą analizę, ale w opcjach MCMC wybierz opcję Sample from prior only. Pozwoli to na wizualizację pełnego rozkładu priorytetu przy braku danych sekwencyjnych. Podsumuj drzewa z pełnego rozkładu priordystrybucji i porównaj podsumowanie z posterior summary tree.
Oszacowanie czasu dywergencji przy użyciu „datowania węzłów” typu opisanego w tym rozdziale zostało zastosowane do odpowiedzi na wiele różnych pytań w ekologii i ewolucji. Na przykład, datowanie węzłów ze skamieniałościami zostało wykorzystane w określaniu różnorodności gatunkowej cykad, analizie tempa ewolucji roślin kwitnących oraz badaniu pochodzenia ciepłych i zimnych pustynnych cyjanobakterii .
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond and Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), nr 1, 114. A Gelman, G Roberts, and W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled i Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, and S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian, and Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith i Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008),no. 5898, 86-89.
Ten dokument został przetłumaczony z LATEX przezHEVEA.