Introduction
Mutacje de novo w linii zarodkowej (DNMs) są zmianami genetycznymi u osobnika spowodowanymi mutagenezą zachodzącą w gametach rodzicielskich podczas oogenezy i spermatogenezy. Termin „de novo” nie powinien być tu mylony z terminem „nowa mutacja”. Pomimo faktu, że DNM w kontekście trio (ojciec, matka i dziecko) są nowymi mutacjami, mogą one być powszechnymi, rzadkimi lub nowymi wariantami w populacji ogólnej. Aby zmierzyć i wyjaśnić częstość występowania danego DNM, należy najpierw ocenić wpływ wariantu na fenotyp, ponieważ nowe korzystne cechy mogą ewoluować, gdy powstające mutacje genetyczne oferują określone korzyści w zakresie przeżycia (Front Line Genomics, 2017).
W ludziach z genetycznymi chorobami niemendlowskimi, które występują sporadycznie, DNM są zwykle nowe, bardziej niezawodne i bardziej szkodliwe niż warianty dziedziczone, ponieważ nie podlegają silnej selekcji naturalnej (Crow, 2000; Front Line Genomics, 2017). Dlatego też identyfikacja genetycznej przyczyny zaburzenia indukowanego przez DNM u danej osoby może stanowić wyzwanie z klinicznego punktu widzenia, ponieważ plejotropia i heterogenność genetyczna mogą leżeć u podstaw pojedynczego fenotypu (Eyre-Walker i Keightley, 2007). W związku z tym, w ostatniej dekadzie podjęto znaczne wysiłki w celu sekwencjonowania eksomów osób z chorobami o niejasnej etiologii genetycznej dla celów diagnostyki klinicznej. Jednakże, nawet po wykryciu kandydujących wariantów de novo, nadal nie ma wystarczających informacji na temat wariantów częstych i rzadkich, co wyklucza jednoznaczne wnioskowanie o patogenności zidentyfikowanego wariantu de novo i jego roli w chorobie (Acuna-Hidalgo i in., 2016). Ograniczenie to można wytłumaczyć faktem, że warianty de novo są zazwyczaj heterozygotyczne i mogą być zarówno niezwykle rzadkie, jak i powszechne. W przypadku bardzo rzadkich wariantów de novo, patogenność wariantu może być trudna do udowodnienia, ponieważ nie ma więcej pacjentów z tym samym fenotypem i wariantem de novo. W przypadku częstych wariantów de novo czynniki decydujące o przejawach patogenności wariantu mogą nie być znane, szczególnie jeśli niektóre osoby w populacji ogólnej mają dany wariant, ale nie mają choroby genetycznej. Jednak niezależnie od tempa powstawania wariantów de novo, oba typy wariantów mogą być skalowane na podstawie względnej sprawności i selekcji naturalnej.
Dostosowalność zależy od wielu czynników; dlatego, aby ocenić, czy DNM jest patogenny lub adaptacyjny, i zrozumieć, dlaczego występuje z określoną częstością w populacji, konieczne jest zbadanie wariantu w odpowiednich warunkach. Obejmują one środowisko, wiek rodziców, kontekst genomowy, epigenetykę i inne czynniki, ponieważ wszystkie one wpływają na wartość średniej względnej sprawności, która wzrasta monotonicznie, podczas gdy siła selekcji maleje (Peck i Waxman, 2018).
Głównym celem tego badania było wyjaśnienie częstości występowania DNM i określenie, jak te mutacje są rozmieszczone w egzomach ogólnej populacji litewskiej. Zbadano również, czy na częstość występowania tych mutacji miał wpływ skład lub parametry strukturalne sekwencji, w których one występowały oraz inne czynniki mogące wpływać na mechanizmy leżące u podstaw powstawania DNM. Wreszcie staraliśmy się ustalić, czy DNM powstały w wyniku intensywnej presji doboru naturalnego na regiony funkcjonalne. Chociaż dystrybucja i intensywność DNMs były przedmiotem wielu badań, nie były one wcześniej badane w populacji litewskiej.
Materiały i Metody
W tym badaniu analizowaliśmy próbki z populacji litewskiej uzyskane w ramach projektu LITGEN (LITGEN, 2011). Zestaw danych składał się z 49 trio, w których łącznie znajdowały się 144 różne osoby. Genomowe DNA ekstrahowano z krwi żylnej metodą fenolowo-chloroformową lub za pomocą automatycznej platformy ekstrakcji DNA TECAN Freedom EVO® (Tecan Schweiz AG, Szwajcaria) w oparciu o metodę cząstek paramagnetycznych. Egzomy sekwencjonowano na systemie sekwencjonowania SOLiD 5500 (75 bp reads). Dane sekwencjonowania przetwarzano i opracowywano za pomocą oprogramowania Lifescope. Egzomy mapowano zgodnie z genomem referencyjnym człowieka build 19. Średnia głębokość odczytu sekwencjonowania wynosiła 38,5. Pliki w formacie BAM matki, ojca i dziecka wygenerowane przez Lifescope zostały połączone za pomocą oprogramowania SAMtools dla każdego trio.
Mutacje de novo zostały zidentyfikowane przez dwa programy: VarScan (Koboldt et al., 2012) oraz VarSeqTM. Potencjalny wariant uznawano za DNM, jeśli został zidentyfikowany u potomstwa, ale nie był obecny w tej samej pozycji u żadnego z rodziców. Ogólnie, 1,752 i 4,756 DNM zostało wykrytych odpowiednio przez VarScan i VarSeqTM. Aby odrzucić fałszywie pozytywne wywołania de novo, gdy nie było wiadomo, czy wszystkie osobniki w trio zostały poprawnie zidentyfikowane, zastosowano konserwatywne filtry na wykrytych parametrach jakości DNM w następujący sposób: (1) jakość genotypu osobnika ≥50; (2) liczba odczytów w każdym miejscu >20. Do zastosowania tych filtrów na danych wygenerowanych przez VarScan użyto oprogramowania SnpSift. Dane wygenerowane przez oprogramowanie VarSeqTM były filtrowane poprzez wybór tych samych parametrów filtrowania w segmencie Trio Workflow. Ponadto, w celu odrzucenia pozostałych wariantów, które były somatyczne (obecne tylko we frakcji sekwencjonowanych komórek krwi) z niską równowagą alleli lub artefaktami sekwencjonowania, DNM były filtrowane poprzez ustawienie progu dla obserwowanej frakcji odczytów u osobników z alternatywnym allelem (równowaga alleli) dla trio (Kong i in., 2012; Besenbacher i in., 2015; Francioli i in., 2015). Dodatkowo, wszystkie możliwe zidentyfikowane i odfiltrowane de novo warianty pojedynczych nukleotydów zostały ręcznie przejrzane przez Integrative Genomics Viewer (Robinson i in., 2011). Ze względu na dużą liczbę zidentyfikowanych DNM, do walidacji wariantów metodą sekwencjonowania Sangera wybrano losowo 51 wariantów de novo pojedynczych nukleotydów. Sekwencjonowanie Sangera przeprowadzono przy użyciu analizatora genetycznego ABI PRISM 3130xl. Wszystkie przefiltrowane i ręcznie zweryfikowane DNM zidentyfikowane przez VarScan (N = 95) i przez VarSeqTM (N = 84) zostały zanotowane przy użyciu ANNOVAR (Butkiewicz i Bush, 2016; Wang i in., 2010). Do analizy interakcji białek wykorzystano oprogramowanie STRING (Szklarczyk i in., 2017). Podobnie jak w przypadku mapowania egzomów, anotacje przeprowadzono z wykorzystaniem referencyjnego genomu ludzkiego hg19.
Prawdopodobieństwo, że pozycja wywołania była DNM w trio, obliczono niezależnie dla każdego trio. Jak opisano w poprzednim odnośniku (Besenbacher i in., 2015), wskaźnik de novo na pozycję na pokolenie (PPPG) obliczono w następujący sposób:
gdzie f jest liczbą trio, a N jest liczbą miejsc możliwych do wywołania, które potencjalnie mogą być zidentyfikowane jako miejsca de novo dla każdego trio oddzielnie, niezależnie od głębokości sekwencjonowania. Liczba ta zmienia się w zależności od trio. ni jest liczbą zidentyfikowanych DNM dla trio i. Prawdopodobieństwo Pji (de novos ingle nucleotide) dla wywołanego pojedynczego miejsca nukleotydowego j i rodziny i, które ma być zmutowane, obliczono w następujący sposób:
Prawdopodobieństwo Pji (de novo indel)dla wywołanego miejsca indelowego j i rodziny i, które ma być zmutowane, obliczono jako:
gdzie C, M, i F oznaczają odpowiednio potomstwo, matkę i ojca, a Hetero, HomR, i HomA oznaczają odpowiednio heterozygotyczny, homozygotyczny dla allelu referencyjnego i homozygotyczny dla allelu alternatywnego. Prawdopodobieństwo Pij (de novo) obliczono w odniesieniu do pokrycia sekwencjonowania. Przedziały ufności dla oszacowań częstości obliczano jak dla proporcji dwumianowych. Do estymacji współczynnika DNM oraz do dalszych obliczeń wykorzystano pakiet R (wersja 3.4.3) (R Core Team, 2013).
W celu przetestowania hipotezy, że różnice w tempie DNM w różnych regionach genomu mogą być wyjaśnione przez wewnętrzne cechy samego regionu genomowego oraz wiek rodzica, przeprowadzono analizę regresji liniowej, dla której przeprowadzono „wtórną” anotację każdego DNM, wykorzystując dane z projektów ENCODE (ENCODE Project Consortium, 2012) i LITGEN (LITGEN, 2011). W pierwszej kolejności, zgodnie z wcześniejszym opracowaniem (Besenbacher i in., 2015), w celu zebrania rekordów dotyczących krajobrazu genomowego zidentyfikowanych DNM, wybrano limfoblastoidalne linie komórkowe (LCL i GM12878) (ENCODE Project Consortium, 2012). Zebrano dane dla:
(1) wskaźników ekspresji (eQTL) (ENCODE Project Consortium, 2012; Lappalainen i wsp., 2013; GTEx Consortium i wsp., 2017) w różnych tkankach. Według ekspresji regiony z DNM zostały podzielone na pozycje o ekspresji specyficznej i niespecyficznej;
(2) pomiary miejsc nadwrażliwości DNase1 (DHS). Status DHS był przypisywany 0, jeśli znajdował się poza szczytem DHS i 1, jeśli w jego obrębie;
(3) pomiary kontekstu wysp CpG. Jeśli DNM znajdował się w obrębie wysp CpG, status pozycji przypisywano 1; jeśli poza – 0;
(4) trzy znaczniki histonowe (H3K27ac, H3K4me1, i H3K4me3) z projektu ENCODE. Jeśli DNM znajdował się w pozycji oznaczonej histonem, przypisywano mu wartość 1, a jeśli nie – 0;
(5) Wartości konserwatywne GERPP++ zostały zebrane przy użyciu narzędzia do anotacji ANNOVAR. Zgodnie z wartościami konserwatywnymi pozycje z DNM zostały podzielone na konserwatywne (GERP++ score >12) i niekonserwatywne (GERP++ score <12) (Davydov i in., 2010; ENCODE Project Consortium, 2012). Na podstawie zapisów ankietowych z projektu LITGEN zebrano dane dotyczące wieku rodziców. Po zebraniu parametrów dla każdego trio obliczono liczbę stanowisk z każdym parametrem. Następnie przeprowadzono analizę korelacji, a następnie modelowanie regresji liniowej DNM i parametrów.
Wyniki
Po analizie DNM, dla dwóch trio (nr 4 i 21) zidentyfikowano wyjątkowo dużą liczbę DNM: 113 i 123 (odpowiednio przez VarScan i VarSeqTM) oraz 16 (VarScan). Wyniki te skłoniły nas do przeprowadzenia testu biologicznego ojcostwa, który został odrzucony dla tria nr 4 i potwierdzony dla tria nr 21. 4 i potwierdzone dla tria nr. 21. W związku z tym dane dla tria nr 4 zostały wyłączone z badań. 4 zostały wyłączone z badania. W ostatecznym zestawie 48 triów, 95 DNM zostało zidentyfikowanych w 34 triach przy użyciu oprogramowania VarScan, a 84 DNM w 31 triach zostały zidentyfikowane przy użyciu oprogramowania VarSeqTM (Ryc. 1). Żadnych DNM nie wykryto w 18 i 15 trio, odpowiednio za pomocą VarScan i VarSeqTM. Spośród wszystkich DNM zidentyfikowanych przez oba programy, tylko 5,37% DNM pasowało do siebie (trzy DNM w genach MEIS2, PGK1 i MT1B). Każda osoba miała średnio 1,9 (oprogramowanie VarScan) i 1,7 (VarSeqTM) DNMs.
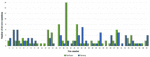
FIGURE 1. Porównanie de novo pojedynczych wariantów nukleotydowych zidentyfikowanych przez oprogramowanie VarScan (niebieski) i VarSeqTM (zielony).
Analiza 95 DNMs, które zostały zidentyfikowane przez oprogramowanie VarScan wykazała, że 20 DNMs było egzonicznych, w tym dwa DNMs stop-gain, siedem synonimicznych DNMs i 11 niesynonimicznych DNMs. Osiemdziesiąt nowych mutacji zidentyfikowanych przez VarSeqTM było egzonicznych, w tym 1 DNM typu stop-gain i 78 niesynonimicznych DNM (Figura 2). Większość DNM zidentyfikowanych przez VarScan znajdowała się w chromosomach 1, 2, 4 i 5, podczas gdy VarSeqTM zidentyfikował DNM głównie w chromosomach 2, 6, 7 i 11. Liczba zidentyfikowanych DNM nie korelowała z gęstością genów w chromosomach (R = 0,09, p-value = 0,65 dla VarScan i R = 6,73, p-value = 0,51 dla VarSeqTM) ani z wielkością chromosomów (Ryc. 3). Według obu programów proporcje tranzycji i transwersji były bardzo podobne: odpowiednio 1,44 i 1,47 (Rysunek 4). Stwierdzono jednak różnice w strukturze przejść. W szczególności, wśród DNM zidentyfikowanych przez VarScan więcej było zmian G/T i A/C, natomiast wśród DNM zidentyfikowanych przez VarSeqTM więcej było zmian A/T i G/C.
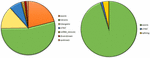
RYSUNEK 2. Skład mutacji de novo (DNMs) generowanych przez VarScan (po lewej) i przez VarSeqTM (po prawej).
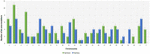
RYSUNEK 3. Rozkład liczby wariantów de novo według chromosomów zgodnie z danymi wygenerowanymi przez VarScan i VarSeqTM. Zielone słupki reprezentują DNMs zidentyfikowane przez oprogramowanie VarScan, niebieskie – przez VarSeqTM. Słupki błędów reprezentują błąd standardowy średniej DNM dla każdego chromosomu.
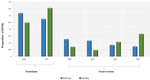
FIGURE 4. Zdarzenia molekularne leżące u podstaw tranzycji występują częściej niż te prowadzące do transwersji, co skutkuje ∼ 1,5-krotnie większą częstością tranzycji w stosunku do transwersji w całym egzomie. Zdarzenia przejścia i transwersji zidentyfikowane przez oprogramowanie VarScan (zielony) i VarSeqTM (niebieski). Słupki błędów reprezentują błąd standardowy średniej DNMs.
Obliczone wskaźniki mutacji de novo pojedynczego nukleotydu wynosiły 2,4 × 10-8 PPPG (95% przedział ufności : 1,96 × 10-8-2,99 × 10-8) według VarSeqTM i 2.74 × 10-8 na nukleotyd na pokolenie (95% CI: 2,24 × 10-8-3,35 × 10-8) według VarScan.
Trzy indele de novo w trzech triach zostały zidentyfikowane przez algorytm VarScan w chromosomach 6 i 11. Obliczony wskaźnik indeli de novo w genomie wynosił 1,77 × 10-8 (95% CI: 6,03 × 10-9-5,2 × 10-8) PPPG. Warto zauważyć, że wszystkie indeksy de novo były „odwracalne”, tj. Rodzice mieli nowe warianty w genomie, a ich dzieci miały warianty de novo oparte na genomie referencyjnym o średniej wartości głębokości sekwencjonowania 37,5 i jakości genotypu 50, odpowiednio. Jednakże, te trzy DNM nie zostały wybrane do walidacji metodą sekwencjonowania Sangera, przez co pozostaje prawdopodobieństwo przeszacowania indeli de novo. De novo indelami były C/T i A/G w kontekście pojedynczych nukleotydów.
Modelowanie regresji liniowej ujawniło, że miejsca nadwrażliwości DNAse 1, kontekst wysp CpG, wartości konserwatywne GERPP++ i poziomy ekspresji wyjaśniały ∼68-93% wskaźników DNM (Tabela 1). Ani markery epigenetyczne, ani wiek ojcowski nie korelowały istotnie z częstością DNM. Modele ustalono tylko na podstawie danych uzyskanych z VarScan, ponieważ nie było korelacji między danymi z VarSeqTM a wewnętrznymi cechami samego regionu genomowego.

TABELA 1. Regresja liniowa miejsc nadwrażliwości DNAaseI, kontekstu wysp CpG, wartości konserwatywnych GERPP++ i wpływu poziomu ekspresji na częstość DNMs.
Funkcjonalna predykcja DNMs
W celu oceny, które mutacje missense były deleteryjne i zmieniały funkcję uszkodzonego białka ze względu na typ, przeanalizowano przewidywane punktacje kategoryczne dla uszkodzeń indukowanych przez DNMs. Pod uwagę wzięto 10 następujących wartości: polifen HDIV i HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, kodowanie Fathmm-MKL i GERP++. Na podstawie przewidywanych wyników wybrano cztery DNM zidentyfikowane przez VarScan jako mające sześć lub więcej szkodliwych lub prawdopodobnie szkodliwych przewidywań. Te DNM typu stop-gain znajdowały się w genach MEIS2 i ULK4, natomiast niesynonimiczne DNM w genach MT1B i PGK1. Białka kodowane przez te geny są ważne dla wzrostu neuronów, endocytozy i ochrony przed negatywnym wpływem metali ciężkich. Białka te uczestniczą w uwalnianiu inhibitora nowotworowych naczyń krwionośnych – angiostatyny oraz w różnych szlakach sygnałowych. Nie stwierdzono żadnych połączeń pomiędzy białkami kodowanymi przez te geny (rysunek 5).
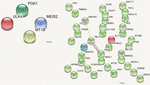
RYSUNEK 5. Interakcje białko-białko (Szklarczyk i in., 2017) w genach będących nosicielami DNM. DNMs zidentyfikowane przez VarScan w genach kodujących białka znajdują się po lewej stronie, DNMs zidentyfikowane przez VarSeqTM – po prawej. Kolorowe linie wskazują na połączenie między białkami.
Mutacje de novo zidentyfikowane przez VarSeqTM były analizowane bardziej szczegółowo, jeśli były przewidywane jako uszkadzające lub prawdopodobnie uszkadzające przez co najmniej połowę narzędzi predykcyjnych. Było 35 mutacji punktowych (patrz ??) w genach kodujących białka, które były ważne dla przebudowy chromatyny, regulacji cytoszkieletu, wzrostu i żywotności komórek, cytoplazmatycznych szlaków sygnałowych oraz inicjacji odpowiedzi neuronalnych wyzwalających percepcję zapachu.
Wśród białek kodowanych przez geny dotknięte DNM, tylko CLPTM1, ZNF547 i DMXL1 były w jakiś sposób połączone (Figura 5).
Dyskusja
W tym badaniu przeprowadziliśmy kompleksową analizę dystrybucji DNM w różnych regionach egzomu w populacji litewskiej. W sumie wykryto 95 DNM w 34 triadach i 84 DNM w 31 triadach przy użyciu technologii sekwencjonowania SOLiD 5500 odpowiednio algorytmami VarScan i VarSeqTM. Na wstępie chcielibyśmy zauważyć, że do wywołania DNMs wybraliśmy VarScan, ponieważ według (Warden i in., 2014) algorytm ten daje w wyniku listę wariantów, o wysokiej zgodności (>97%) do wysokiej jakości wariantów wywołanych przez GATK UnifiedGenotyper i HaplotypeCaller. Oprogramowanie VarSeqTM zostało wybrane, ponieważ jest to szeroko stosowane narzędzie do analizy wariantowej zarówno w badaniach naukowych, jak i analizach klinicznych. Pomimo, że oba algorytmy zostały zaprojektowane do poszukiwania DNM w egzomie potomstwa, które nie były obecne u żadnego z rodziców, zgodność pomiędzy dwoma programami do analizy DNM wyniosła tylko 5,37%. Algorytm VarScan miał wyższą czułość (5,42%) w wykrywaniu DNM przed filtracją niż algorytm VarSeqTM (1,77%). Podejrzewamy więc, że żadne z narzędzi nie zdołało wywołać mutacji z powodu wysokiej czułości, której zawsze towarzyszyła niska specyficzność. Dlatego sugerujemy, że znaczna poprawa wyników może zostać osiągnięta poprzez połączenie danych wyjściowych różnych narzędzi (Sandmann i in., 2017).
Na podstawie wygenerowanych danych oszacowany wskaźnik DNM pojedynczego nukleotydu wynosił od 2,4 × 10-8 do 2,74 × 10-8, a wskaźnik indeli de novo wynosił 1,77 × 10-8 PPPG, w zależności od zastosowanego algorytmu. Obliczony przez nas wskaźnik DNM był wyższy niż raportowany w poprzednich badaniach (Kong i in., 2010, 2012; Neale i in., 2012; Szamecz i in., 2014; Besenbacher i in., 2015; Francioli i in., 2015), w których wahał się między 1,2 × 10-8 a 1,5 × 10-8 PPPG. Wyższy wskaźnik DNM w naszym badaniu był uzasadniony, ponieważ nasze badanie opierało się na danych egzomowych. Dodatkowo, egzomy wykazują znacznie wyższe (o 30%) wskaźniki mutacji niż całe genomy, ponieważ skład par zasad całego genomu różni się od składu egzomów. W szczególności, eksomy mają średnią zawartość GC wynoszącą około 50%, podczas gdy cały genom ma około 40% (Neale i in., 2012). Zmetylowane CpGs reprezentują u ludzi sekwencje wysoce mutowalne ze względu na spontaniczną deaminację zasad cytozyny (Neale i in., 2012). Zgodnie z badaniami genomiki porównawczej, uważa się, że zwiększone tempo mutacji w regionach bogatych w CpG wyewoluowało około czasu radiacji ssaków (Francioli i in., 2015). Podczas dywergencji gatunków, bogate w CpG regiony egzoniczne przeszły zwiększone tempo mutacji w porównaniu do tych w niekodującym DNA i przekształciły się w regiony niekodujące. W związku z tym, wówczas efekt zawartości CpG zmniejsza się z czasem, średnia szybkość mutacji spada aż do osiągnięcia poziomu obecnego w otaczającym niekodującym DNA (Subramanian i Kumar, 2003). Jednakże, podczas gdy sekwencje w neutralnie ewoluujących regionach genomu miały wystarczająco dużo czasu, aby wyrównać się w odniesieniu do kontekstów dinukleotydowych, selekcja oczyszczająca utrzymała hipermutowalne CpGs w regionach funkcjonalnych (Subramanian i Kumar, 2003; Schmidt i in., 2008; Francioli i in., 2015). Dlatego też, ponieważ stwierdziliśmy wyższy wskaźnik DNM niż ten raportowany przez inne badania, spekulowaliśmy, że może to być przynajmniej częściowo spowodowane lokalnym kontekstem sekwencji i/lub możliwą presją selekcji naturalnej na egzom. W związku z tym zastosowano model regresji liniowej i stwierdzono, że nadwrażliwość na DNAse 1, kontekst wysp CpG, wartości konserwatywne GERPP++ i poziom ekspresji wyjaśniają ∼ 68-93% wskaźnika DNM. Wyniki te wskazują, że DNM w egzomie powstawały niezależnie od zachowania sekwencji DNA. Jednakże wskaźnik DNM był wyższy w genach, których produkty były niespecyficzne i w aktywnych transkrypcyjnie regionach promotoropodobnych.
W przeciwieństwie do wyników innych badań (Wong i in., 2016; Sandmann i in., 2017), stwierdziliśmy, że wiek ojcowski nie korelował z wskaźnikiem DNM. Wyniki te można wyjaśnić faktem, że zbiór danych składał się z trio o podobnym wieku rodziców i że analizowano tylko niewielką część (∼1,5%) całego genomu. Na podstawie tych parametrów każda osoba miała średnio tylko 1,9 (VarScan) lub 1,7 (VarSeqTM) DNM w porównaniu do 40-82 w całym genomie (Crow, 2000; Branciamore i in., 2010; Kong i in., 2012; Neale i in., 2012; Besenbacher i in., 2015; Francioli i in., 2015; Wong i in, 2016), podczas gdy liczba indeli de novo w sekwencji kodującej była podobna do tej zidentyfikowanej w (Front Line Genomics, 2017).
Wyniki naszej obszernej analizy funkcjonalnej adnotacji ujawniły, że spośród wszystkich zidentyfikowanych DNM, 4 (VarScan) i 35 (VarSeqTM) warianty były prawdopodobnie patogennymi DNM. Różnica w liczbie patogennych DNM może wynikać z faktu, że w zależności od algorytmu użytego do identyfikacji DNM, udział DNM w sekwencjach kodujących różnił się znacząco. Na przykład, 21,05% DNM zidentyfikowanych przez oprogramowanie VarScan było egzonicznych, podczas gdy 95,24% DNM zidentyfikowanych przez oprogramowanie VarSeqTM było egzonicznych. Te patogenne DNM występowały w genach kodujących białka istotne dla modelowania chromatyny, regulacji cytoszkieletu, modulacji wzrostu i żywotności komórek, funkcji cytoplazmatycznych szlaków sygnałowych oraz inicjacji odpowiedzi neuronalnej. Pomimo tych DNM uznanych za patogenne, wszystkie osoby biorące udział w ankiecie określiły się jako genetycznie „zdrowe”. Wynik ten wskazuje zatem, że pomimo przypuszczalnej patogenności DNM, genomy, w których DNM były zlokalizowane, najwyraźniej tolerowały takie zmiany, tak że manifestacje chorobowe często nie były wyraźne. Według Szamecz i wsp. (2014), im częściej DNM występują w konserwatywnych pozycjach genetycznych, tym silniejsze są efekty działania doboru naturalnego na zmiany genetyczne poprzez kompensacyjne mechanizmy ochrony genomu. Szkodliwy wpływ wariantów może być łagodzony na cztery sposoby. Niektóre geny mogą tolerować obcięte warianty białek, ponieważ ich efekty funkcjonalne są maskowane przez niepełną ekspresję, warianty kompensacyjne lub niskie znaczenie funkcjonalne obcięcia (Bartha i in., 2015). Z kolei zmiany w genach związane z niesynonimicznymi DNM są kompensowane poprzez mechanizm użytecznej akumulacji mutacji w całym genomie (Szamecz i in., 2014). Sugeruje to, że w tych przypadkach mutacje patogenne nie są na tyle deleteryjne, aby obniżyć średnią kondycję i dlatego utrzymują się dłużej w wielu pokoleniach będąc kształtowane przez dobór naturalny.
Podsumowując, nasza analiza dystrybucji DNMs oraz ich kontekstu genetycznego i epigenetycznego dostarczyła wglądu w zmienność genetyczną genomu litewskiego. W oparciu o te wyniki, dodatkowe badania w grupach pacjentów z chorobami genetycznymi mogą ułatwić naszą zdolność do odróżnienia niektórych patogennych DNM od tolerowanych DNM tła i zidentyfikowania wiarygodnych przyczyn DNM. Głównym ograniczeniem tego badania było jednak to, że nie badaliśmy zmienności w niekodujących i regulacyjnych regionach genów. Informacje te mogłyby przyczynić się do wyjaśnienia możliwych mechanizmów powstawania DNM, które wciąż pozostają niewystarczająco jasne.
Kody akcesyjne
Dane sekwencji zostały zdeponowane w Europejskim Archiwum Nukleotydów (ENA), pod akcesją PRJEB25864 (ERP107829).
Oświadczenie etyczne
Badanie to zostało przeprowadzone zgodnie z zaleceniami pozwolenia, Vilnius Regional Ethics Committee for Biomedical Research. Protokół został zatwierdzony przez Wileński Regionalny Komitet Etyczny do Badań Biomedycznych. Wszyscy badani wyrazili pisemną świadomą zgodę zgodnie z Deklaracją Helsińską.
Author Contributions
LP przeprowadził analizę danych i przygotował manuskrypt. AJ obliczył odsetek mutacji de novo. Sekwencjonowanie triowych eksomów zostało wykonane przez LA i IK. VK był głównym badaczem.
Funding
Ta praca była wspierana przez Europejski Fundusz Społeczny w ramach działania Global Grant. Projekt LITGEN nr. VP1-3.1-ŠMM-07-K-01-013.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Materiały uzupełniające
Materiały uzupełniające do tego artykułu można znaleźć online pod adresem: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). The characteristics of heterozygous protein truncating variants in the human genome. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). In silico functional annotation of genomic variation. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Pochodzenie, wzorce i implikacje ludzkich spontanicznych mutacji. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., and Keightley, P. D. (2007). The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics Magazine Issue 14 – ASHG. London: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Genetyczny wpływ na ekspresję genów w ludzkich tkankach. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., 't Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Dostępne pod adresem: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Czym jest adaptacja i jak należy ją mierzyć? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). A Language and Environment for Statistical Computing. Wiedeń: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., and Sunyaev, S. (2008). Hypermutable non-synonymous sites are under stronger negative selection. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Neutralne substytucje występują w szybszym tempie w eksonach niż w niekodującym DNA w genomach naczelnych. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). The genomic landscape of compensatory evolution Be. The genomic landscape of compensatory evolution (Genomiczny krajobraz ewolucji kompensacyjnej). PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Detailed comparison of two popular variant calling packages for exome and targeted exon studies. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Nowe obserwacje na temat wpływu wieku matki na germline de novo mutacji. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar
.