Alexei Drummond, Andrew Rambaut, Remco Bouckaert, e Walter Xie
1 Introdução
Este tutorial introduz o software BEAST para análise evolutiva Bayesiana através de um simples tutorial. O tutorial envolve a co-estimativa de uma filogenia genética e tempos de divergência associados na presença de informações de calibração a partir de evidências fósseis.
Você precisará do seguinte software à sua disposição:
- BEAST – este pacote contém o programa BEAST, BEAUti, TreeAnnotator e outros programas utilitários. Este tutorial é escrito para BEAST v2.2.x, que tem suporte para múltiplas partições. Ele está disponível para download em http://www.beast2.org/.
- Tracer – este programa é usado para explorar a saída do BEAST (e outros programas MCMC Bayesianos). Ele resume gráfica e quantitativamente as distribuições de parâmetros contínuos e fornece informações de diagnóstico. No momento da escrita, a versão atual é a v1.6. Está disponível para download em
http://tree.bio.ed.ac.uk/software/. - FigTree – esta é uma aplicação para exibir e imprimir filogenias moleculares, em particular aquelas obtidas usandoBEAST. No momento da escrita, a versão atual é a v1.4.2. Ela está disponível para download em http://tree.bio.ed.ac.uk/software/.
Este tutorial irá guiá-lo através da análise de um alinhamento de sequências amostradas de doze espécies de primatas (ver Figura 1). O objetivo é estimar a filogenia, a taxa de evolução em cada linhagem e as idades das divergências astrais não-calibradas.
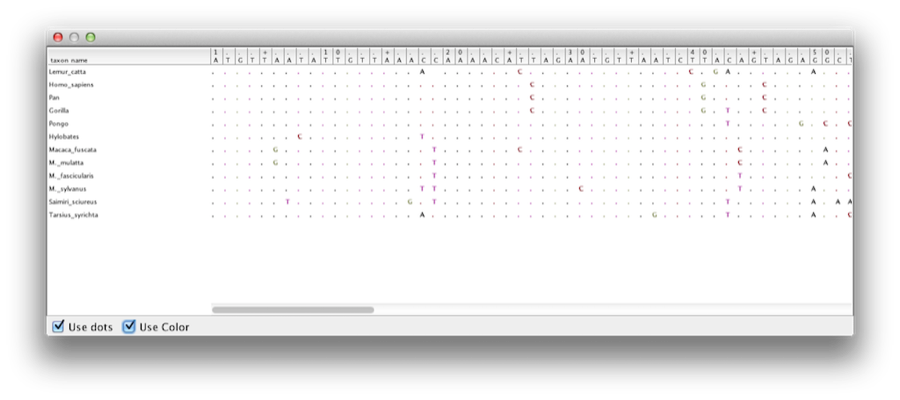
Figure 1: Parte do alinhamento para primatas.
O primeiro passo será converter um arquivo NEXUS com um bloco de DADOS ou CARACTERES em um arquivo de entrada BEAST XML. Isto é feito usando o programa BEAUti (que significa Bayesian Evolutionary Analysis Utility). Este é um programa de fácil utilização para definir o modelo evolutivo e opções para a análise MCMC. O segundo passo é realmente executar BEAST usando o arquivo de entrada gerado pelo BEAUTi, que contém os dados, modelo e configurações de análise. O passo final é explorar a saída do BEAST a fim de diagnosticar problemas e resumir os resultados.
2 BEAUti
O programa BEAUti é um programa de fácil utilização para definir os parâmetros do modelo BEAST. Execute o BEAUti clicando duas vezes sobre o seu ícone. Uma vez executado, o BEAUti terá um aspecto semelhante, independentemente do sistema informático em que estiver a correr. Para este tutorial, a versão Mac OS X é usada nas figuras mas as versões Linux e Windows terão o mesmo layout e funcionalidade.
2.1 Carregando o arquivo NEXUS
Para carregar um alinhamento do formato NEXUS, basta selecionar a opção Importar Alinhamento… do menu File, ou arrastar o arquivo para o meio do painel Partições.
O arquivo de exemplo chamado primate-mtDNA.nex está disponível no diretório examples/nexus/ para Mac e Linux e examples/nexus/ para Windows dentro do diretório onde BEAST foi instalado. Este arquivo contém um alinhamento de sequências de 12 espécies de primatas.
Uma janela Add Partition (Figura 2) apareceria se o pacote relacionado fosse instalado. Se você estiver usando BEAST 2 “puro”, você pode ir para o próximo parágrafo. Caso contrário, selecione Add Alignment e clique em OK para continuar.
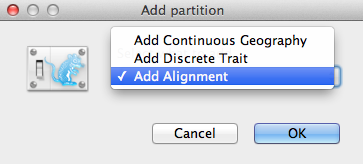
Figure 2: Add Partition window (Somente aparecerá se os pacotes relacionados estiverem instalados).
Se houver alguma sobreposição de codificação nas partições, a janela de mensagem de aviso (Figura 3) aparecerá. Leia e clique em OK para continuar.
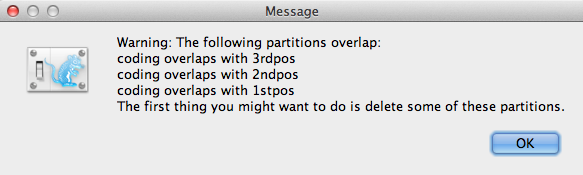
Figure 3: Janela de mensagem de aviso (Só aparece se houver alguma sobreposição de codificação nas partições).
Onça carregada, as partições de cinco caracteres são exibidas no painel principal (Figura 4). O alinhamento é dividido em uma parte codificadora de proteínas e uma parte não codificadora, e a parte codificadora é dividida nas posições de códon 1, 2 e 3. Você deve remover a partição ‘codificadora’ antes de continuar para o próximo passo, pois ela se refere aos mesmos nucleotídeos das partições ‘1stpos’, ‘2ndpos’ e ‘3rdpos’. Para remover a partição ‘codificada’ seleccione a linha e clique no botão ‘-‘ na parte inferior da tabela. Você pode ver o alinhamento clicando duas vezes na partição.
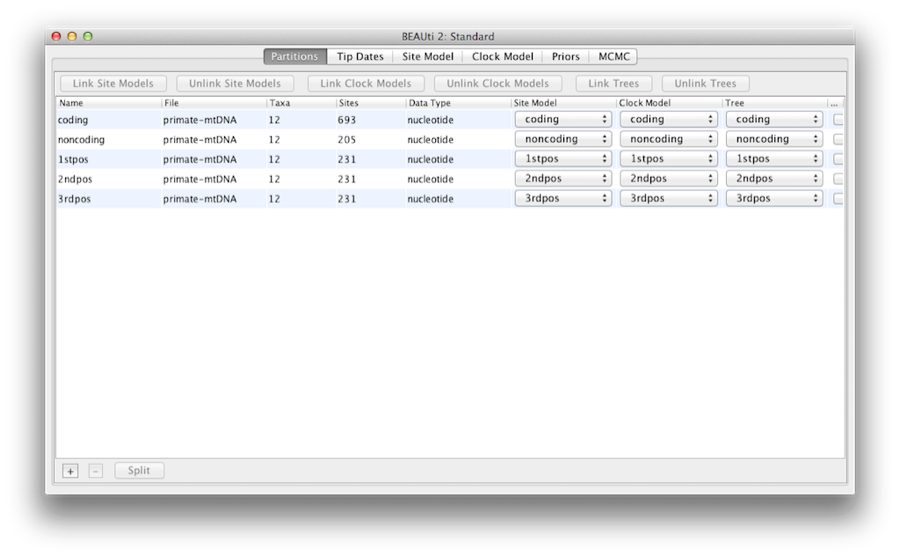
Figure 4: Uma imagem da guia de dados no BEAUti. Esta e todas as seguintes capturas de tela foram feitas em um computador Apple rodando Mac OS X e terão um aspecto ligeiramente diferente em outros sistemas operacionais.
Link/Unlink modelos de partição
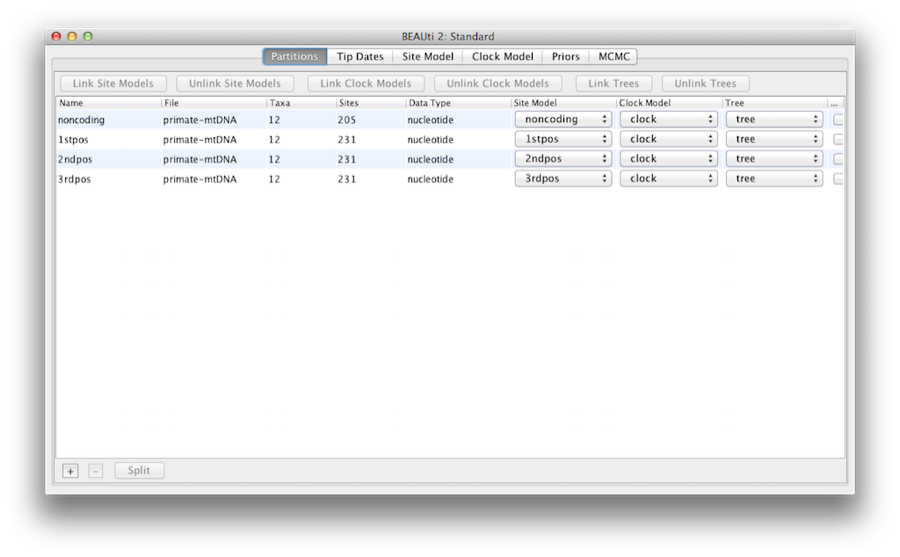
Figure 5: Uma captura de tela da aba Partições no BEAUti depois de ligar e renomear o modelo e a árvore do relógio.
Desde que as sequências estejam ligadas (ou seja, são todas do genoma mitocondrial, que não se acredita ser recombinado em pássaros e mamíferos) partilham a mesma ancestralidade, pelo que as partições devem partilhar a mesma árvore de tempo no modelo. Por uma questão de simplicidade, vamos também assumir que as partições partilham a mesma taxa evolutiva para cada ramo e, portanto, o mesmo “modelo de relógio”. Vamos restringir a nossa modelagem de heterogeneidade de taxas à heterogeneidade do local de cada partição e também permitir que as partições tenham diferentes taxas médias de evolução.
Então, neste ponto vamos precisar ligar o modelo de relógio e a árvore. No painel Partições, selecione todas as quatro partições da tabela (ou nenhuma, por padrão todas as partições são afetadas) e clique no botão Link Trees e depois no botão Link Clock Models (veja a Figura 5). Em seguida, clique no primeiro menu suspenso na coluna Modelo de Relógio e renomeie o modelo de relógio compartilhado para ‘relógio’. Da mesma forma, renomeie a árvore compartilhada para ‘árvore’. Isto tornará as seguintes opções e arquivos de log gerados mais fáceis de ler.
2.2 Configurando o modelo de substituição
O próximo passo é configurar o modelo de substituição. Em seguida, selecione a aba Site Models no topo da janela principal (saltamos a aba Datas de Dicas já que todos os taxa são de amostras contemporâneas). Isto irá revelar as configurações do modelo evolutivo para BEAST. As opções disponíveis dependem se os dados são nucleotídeos, ou aminoácidos, dados binários, ou dados gerais. As configurações que aparecerão após o carregamento do alinhamento dos nucleotídeos primatas serão os valores padrão para os dados dos nucleotídeos, então precisamos fazer algumas alterações.
Figure 6: Uma captura de tela da guia do modelo do site em BEAUti.
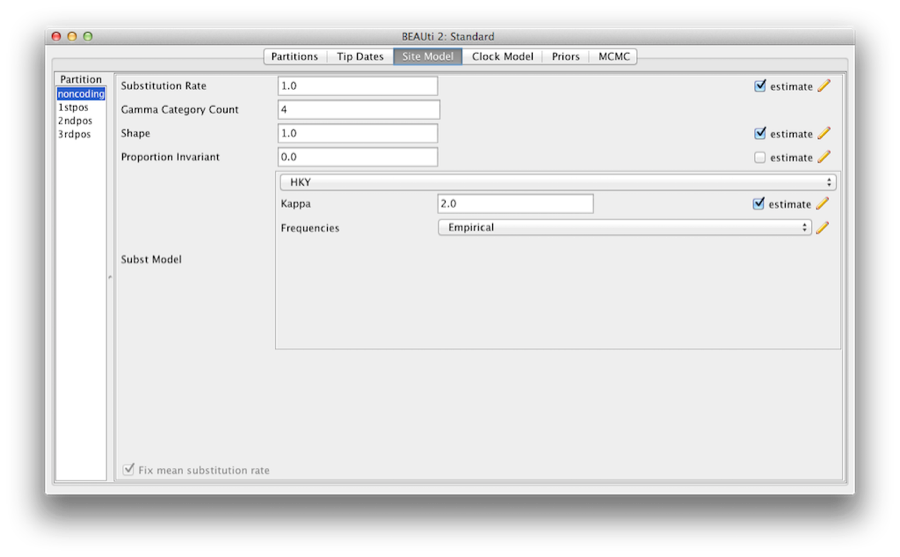
A maior parte dos modelos deve ser familiar para você. Primeiro, defina a Contagem de Categoria Gama para 4 e depois marque a caixa ‘estimativa’ para o parâmetro Forma. Isto permitirá a variação da taxa entre locais em cada partição a ser modelada. Note que 4 a 6 categorias funcionam suficientemente bem para a maioria dos conjuntos de dados, enquanto que ter mais categorias leva mais tempo a calcular para pouco benefício adicional. Deixamos a entrada Proporção Invariante definida como zero.
Então selecione HKY no menu suspenso Subst Modelo. Idealmente, um modelo de substituição deve ser selecionado que se encaixe melhor nos dados de cada partição, mas aqui por uma questão de simplicidade usamos HKY para todas as partições. Além disso, selecione Empirical no menu suspenso Frequências. Isto irá fixar as frequências para as proporções observadas nos dados (para cada partição individualmente, uma vez que desvinculamos os modelos do site). Esta abordagem significa que podemos obter um bom ajuste para os dados sem estimar explicitamente estes parâmetros. Nós fazemos isso aqui simplesmente para tornar os arquivos de log um pouco mais curtos e mais legíveis em partes posteriores do exercício.
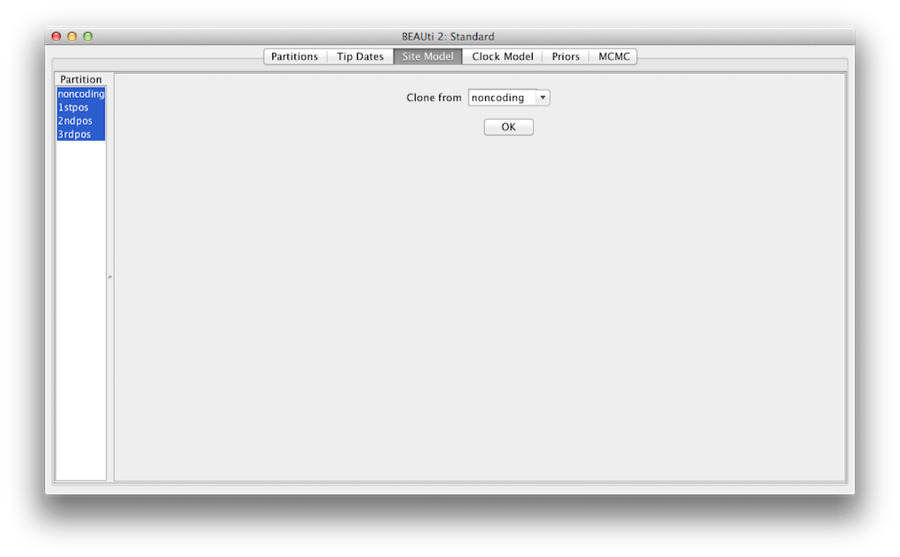
Figure 7: clone configuração de um modelo do site para outros.
>
Sete a caixa ‘estimate’ para o parâmetro Taxa de substituição e selecione a caixa de seleção Fix mean mutation rate. Isso permitirá que as partições individuais tenham suas taxas relativas estimadas para os modelos de locais desvinculados (Figura 6).
Finalmente, segure a tecla ‘shift’ para selecionar todos os modelos de locais no lado esquerdo, e clique em OK para clonar a configuração de não-codificação para 1stpos, 2ndpos e 3rdpos (Figura 7). Percorra cada modelo de site, como você pode ver, suas configurações são as mesmas agora.
2.3 Configurando o modelo do relógio
O próximo passo é selecionar a aba Modelos do Relógio no topo da janela principal. Aqui é onde selecionamos o modelo de relógio molecular. Para este exercício vamos deixar a seleção no valor padrão de um relógio molecular rígido, porque estes dados são muito parecidos com um relógio, e não precisa de variação de taxa entre ramos para ser incluído no modelo.
Para testar a semelhança do relógio, você pode (i) executar a análise com um modelo de relógio relaxado e verificar quanta variação entre taxas está implícita nos dados (veja coeficiente de variação para mais informações sobre isso), ou (ii) executar uma comparação de modelo entre um relógio estrito e relaxado usando amostragem de caminhos, ou (iii) usar um modelo de relógio local aleatório que considera explicitamente se cada ramo da árvore precisa de sua própria taxa de ramificação.
2.4 Priors
A aba Priors permite que os priors sejam especificados para cada parâmetro no modelo deles. As seleções de modelos feitas no modelo do site e nas abas do modelo do relógio, resultam na inclusão de vários parâmetros no modelo, e estes são mostrados na aba Priors (ver Figura 8).
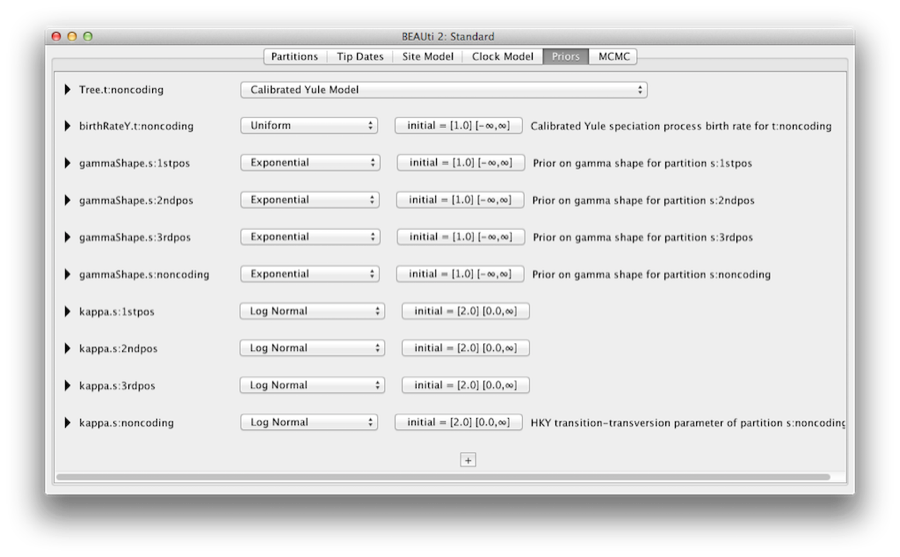
Figure 8: Uma captura de tela da aba Priors no BEAUti.
Aqui também especificamos que queremos usar o modelo Yule calibrado como a árvore anterior. O modelo de Yule é um modelo simples de especiação que é geralmente mais apropriado quando consideramos seqüências de espécies diferentes. Selecione o modelo Yule calibrado no menu suspenso Árvore anterior.
2.4.1 Definindo o nó de calibração
Agora precisamos especificar uma distribuição prévia no nó calibrado, com base em nosso conhecimento fóssil. Isto é conhecido como calibração da nossa árvore. Para definir um anterior extra, pressione o pequeno + botão abaixo da lista de antecedentes. Se não estiver visível em sua visualização, por favor desça o painel até o fundo para encontrar o botão +. Você verá um diálogo que lhe permite definir um subconjunto dos taxa na árvore filogenética. Uma vez criado um conjunto de táxons, você poderá adicionar mais tarde a informação de calibração para o seu mais recente “commonancestor” (MRCA).
Nomear o conjunto de táxons preenchendo a entrada da etiqueta do conjunto de táxons. Chame-o de Human-chimp, já que ele conterá os taxa para Homo sapiens e Pan. Na lista abaixo você verá os táxons disponíveis. Selecione cada um dos dois taxa por vez e pressione o botão de seta >>. Como este é um nó calibrado para ser usado em conjunto com o Yule calibrado anteriormente, a monofilia deve ser aplicada, portanto selecione a caixa de seleção marcada Monofilética. Isto irá restringir a topologia da árvore para que o agrupamento homem-chimpanzé seja mantido monofilético durante o curso da análise MCMC.
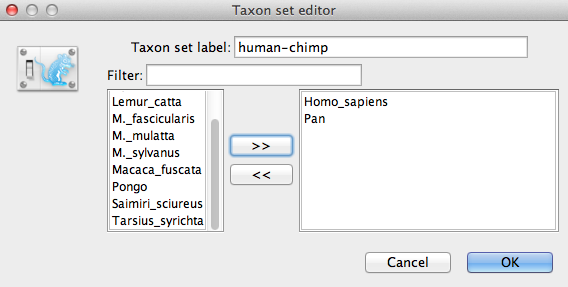
Figure 9: Taxon set editor no BEAUti.
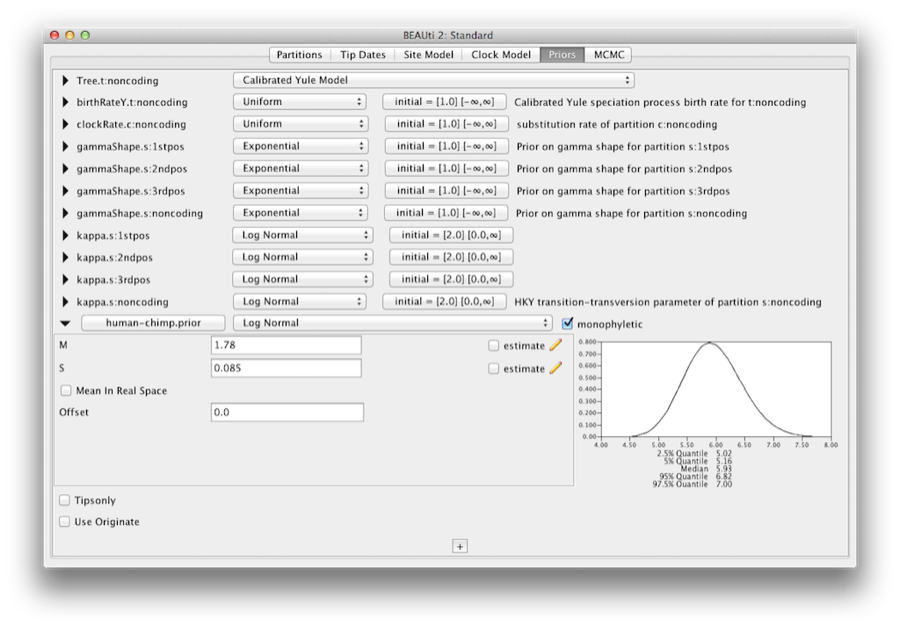
Para codificar as informações de calibração, precisamos especificar uma distribuição para o MRCA do human-chimp.com. Selecione a distribuição Log-normal no menu suspenso à direita do human-chimp.prior recém-adicionado. Clique no triângulo preto e aparecerá um gráfico da função de densidade de probabilidade, juntamente com os parâmetros para a distribuição normal do log. Vamos definir M=1,78 e S=0,085 que especificará uma distribuição centrada em cerca de 6 milhões de anos com um desvio padrão de cerca de 0,5 milhões de anos. Isto dará um intervalo central de probabilidade de 95% cobrindo 5-7 Mya. Isto corresponde aproximadamente à estimativa atual da data do mais recente ancestral comum dos humanos e chimpanzés (Figura 10).
Figure 10: Uma captura de tela das opções prévias de calibração no painel de Priores no BEAUti.
Devemos convencer-nos de que os priores apresentados no painel de Priores reflectem realmente a informação prévia que temos sobre os parâmetros do modelo. Finalmente especificaremos também alguns priores difusos “não-informativos”, mas adequados, sobre a taxa de relógio molecular global (clockRate) e a taxa de especiação (birthRateY) da árvore Yule anterior. Para cada um deles, selecione Gama no menu suspenso e, usando o botão de seta, expanda a visualização para revelar os parâmetros do anterior do Gama. Tanto para a taxa de relógio quanto para a taxa de nascimento do Yule, ajuste o parâmetro Alfa(forma) para 0,001 e o parâmetro Beta (escala) para 1000 (Figura 11).
Por padrão, cada um dos parâmetros de forma gama tem uma priordistribuição exponencial com uma média de 1. Isso implica (veja a Figura 3.7) que esperamos uma variação somada. Por padrão os parâmetros kappa para o modelo HKY têm uma distribuição prévia lognormal(1,1.25), que concorda amplamente com a evidência empírica sobre o intervalo de valores realistas para asbias de transição/transversão. Estes priores por defeito são mantidos uma vez que são adequados para esta análise particular.
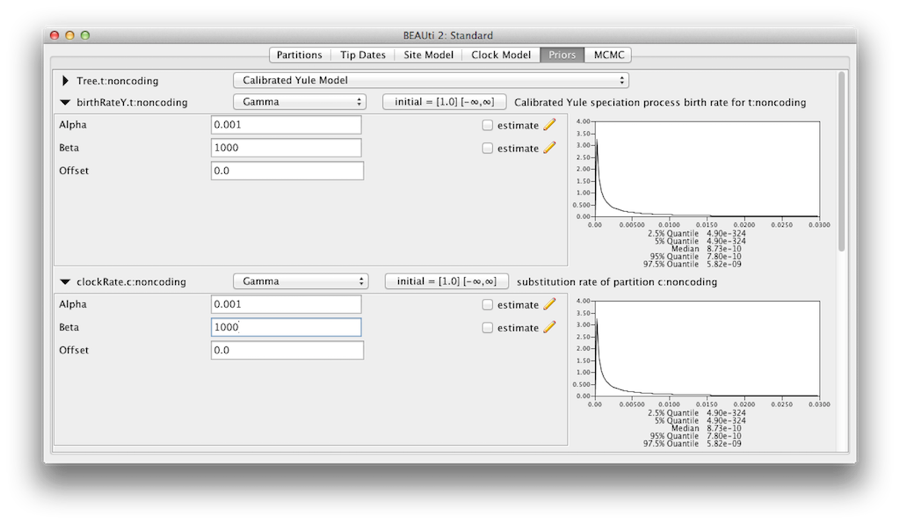
Figure 11: Gamma prior.
2.5 Definindo as opções MCMC
O separador seguinte, MCMC, fornece mais configurações gerais para controlar o comprimento da execução do MCMC e os nomes dos ficheiros.
Primeiro temos o Comprimento da Corrente. Este é o número de passos que o MCMC fará na cadeia antes de terminar. Quanto tempo isto deve ser depende do tamanho do conjunto de dados, da complexidade do modelo deles e da qualidade da resposta requerida. O valor padrão de 10.000.000 é inteiramente arbitrário e deve ser ajustado de acordo com o tamanho de seu conjunto de dados. Para este conjunto de dados vamos definir o comprimento da corrente para 6.000.000, já que isso funcionará razoavelmente rápido na maioria dos moderncomputadores (alguns minutos).
O Armazenamento Cada campo determina com que frequência o estado é armazenado para arquivar. Armazenar o estado periodicamente é útil para situações em que o ambiente de computação não é muito confiável e uma execução BEAST pode ser interrompida. O campo Pre Burnin especifica o número de amostras que não estão registradas logo no início da análise. Deixamos os campos Store Every e Pre Burnin definidos para os seus valores padrão. Abaixo destes estão os detalhes dos arquivos de log. Cada um pode ser expandido clicando no triângulo preto.
As opções seguintes especificam com que frequência os valores dos parâmetros no Markovchain devem ser mostrados na tela e gravados no arquivo de log.A saída da tela é simplesmente para monitorar o progresso dos programas socan ser definido para qualquer valor (embora se definido muito pequeno, a quantidade de informação que está sendo exibida na tela irá realmente retardar o programa). Para o arquivo de log, o valor deve ser definido em relação ao comprimento total da cadeia. A amostragem com muita freqüência resultará em arquivos grandes invertidos com pouco benefício extra em termos de precisão da análise. A amostragem é muito rara e o arquivo de log não registrará informações suficientes sobre as distribuições dos parâmetros. Você provavelmente quer ter como objetivo armazenar não mais do que 10.000 amostras, então isto deve ter um comprimento de cadeia não inferior a 10.000.
Para este exercício, vamos definir o log de rastreamento e a freqüência do log da árvore para 1.000 e o log da tela para 10.000. Especifique também Primates.log como o nome do arquivo de log de rastreamento e Primates.trees como o nome do arquivo de log da árvore.Certifique-se de que o nome do arquivo arquivado do log da tela esteja vazio, ou o log da tela não será escrito na tela.
- Se você estiver usando o sistema operacional Windows, então sugerimos que você adicione o sufixo .txt a ambos (assim,Primates.log.txt e Primates.trees.txt) para que o Windows reconheça estes arquivos como arquivos de texto.
2.6 Gerando o ficheiro BEAST XML
Estamos agora prontos para criar o ficheiro BEAST XML. Para fazer isso, selecione a opção Salvar no menu Arquivo. Verifique os priors padrão, e salve o arquivo com um nome apropriado (normalmente terminamos o nome do arquivo com .xml, ou seja, Primates.xml).
3 Executando BEAST
Figure 12: Uma captura de tela de BEAST.
Agora execute BEAST e quando ele pedir um arquivo de entrada, forneça seu arquivo XML recém-criado como entrada. BEAST irá então correr até que tenha terminado de reportar a informação para a tela. Os arquivos de resultados reais são salvos no disco no mesmo local que o seu arquivo de entrada. A saída para o ecrã irá aparecer algo parecido com isto:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Note que existe alguma informação útil no início em relação aos alinhamentos e quais as probabilidades de árvore que são usadas. Além disso, todas as citações relevantes para a análise são mencionadas no início da execução, que pode ser facilmente copiada tomanuscripts relatando sobre a análise. Em seguida, segue-se o relato da cadeia, que dá algum feedback em tempo real sobre o progresso da cadeia.
No final, é impressa uma análise do operador, que lista todos os operadores utilizados na análise, juntamente com a freqüência com que o operador foi tentado, aceito e rejeitado (ver colunas #total, #aceito e #rejeitado, respectivamente). A taxa de aceitação é a proporção de vezes que um operador é aceito quando ele é selecionado para fazer uma proposta. Em geral, uma taxa de aceitação elevada, digamos acima de 0,5 indica que as propostas são conservadoras e não exploram o espaço de parâmetros de forma eficiente. Por outro lado, uma taxa de aceitação baixa indica que as propostas são demasiado agressivas e quase sempre resultam num estado que é rejeitado por causa do seu posterior baixo. Uma taxa de aceitação de 0,234 é o alvo (baseado em evidências muito limitadas fornecidas por ) para muitos (mas não todos) operadores implementados em BEAST.
Alguns operadores têm um parâmetro de ajuste, por exemplo o fator de escala do parâmetro ascale. Se a taxa de aceitação final não estiver próxima do alvo, BEAST irá sugerir um novo valor para o parâmetro de sintonia, que é impresso na análise do operador. Neste caso, todas as taxas de aceitação são boas para os operadores que têm parâmetros de sintonia. Os operadores sem parâmetros de ajuste incluem os operadores wideexchange e Wilson-Balding para esta análise. Ambos os operadores tentam mudar a topologia da árvore com grandes passos, mas como os dados suportam uma única topologia esmagadoramente, estas propostas radicais são quase sempre rejeitadas.
4 Analisando os resultados
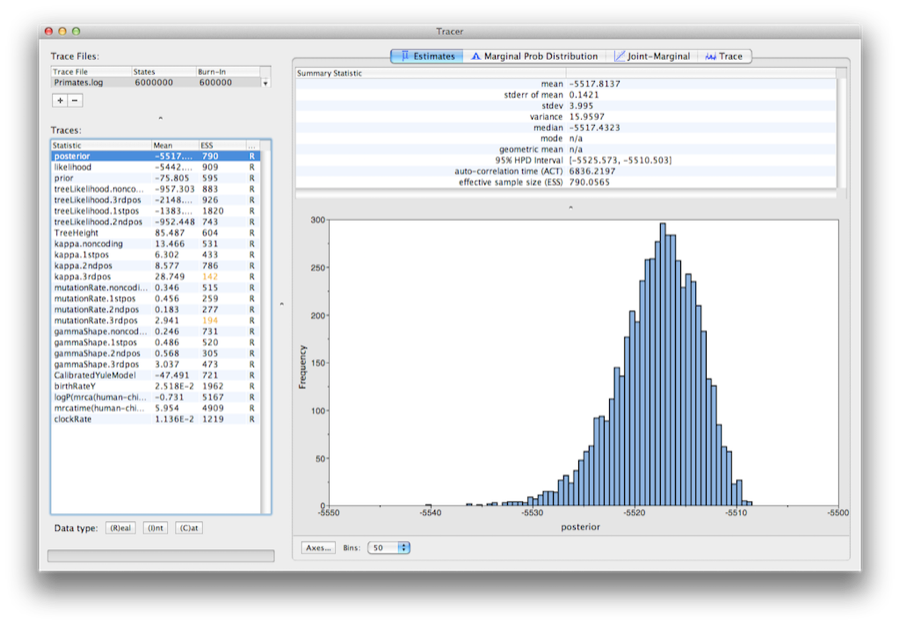
Figure 13: Uma captura de tela do Tracer v1.6.
Executar o programa chamado Tracer para analisar a saída do BEAST. Quando a janela principal estiver aberta, escolha Importar arquivo de rastreamento… no menu File e selecione o arquivo queBEAST criou chamado Primates.log (Figura 13).
Lembrar que o MCMC é um algoritmo estocástico, então os números reais não serão exatamente os mesmos que os representados na figura.
No lado esquerdo há uma lista das diferentes quantidades que o BEAST registrou no arquivo. Existem traços para o posterior (este é o logaritmo natural do produto da probabilidade da árvore e a densidade anterior), e os parâmetros contínuos. Selecionando um traço a esquerda traz análises para este traço no lado direito, dependendo da aba que for selecionada. Quando aberto pela primeira vez, o ‘traço posterior’ é selecionado e várias estatísticas deste traço são mostradas na aba Estimativas. No canto superior direito da janela há uma tabela de estatísticas calculadas para o traço selecionado.
Selecionar o parâmetro clockRate na lista da esquerda para ver a taxa média de evolução (média sobre toda a árvore e todos os sites). O traçador irá traçar um histograma (marginal posterior) para a estatística selecionada e também lhe dará estatísticas resumidas como a média e a mediana. O HPD 95% representa o maior intervalo de densidade posterior e representa o intervalo mais compacto no parâmetro selecionado que contém 95% da probabilidade posterior. Pode ser pensado vagamente como um análogo Bayesiano a um intervalo de confiança. O parâmetro TreeHeight dá a distribuição marginal posterior da idade da raiz da árvore inteira.
Selecione o parâmetro TreeHeight e depois Ctrl-clique mrcatime(human-chimp) (Command-clique no Mac OS X). Isto mostrará uma exibição da idade da raiz e da calibração MRCA que especificamos anteriormente no BEAUti. Você pode verificar que a divergência que usamos para calibrar a árvore(mrcatime(human-chimp)) tem uma distribuição posterior que corresponde à distribuição anterior que especificamos (Figura 14).
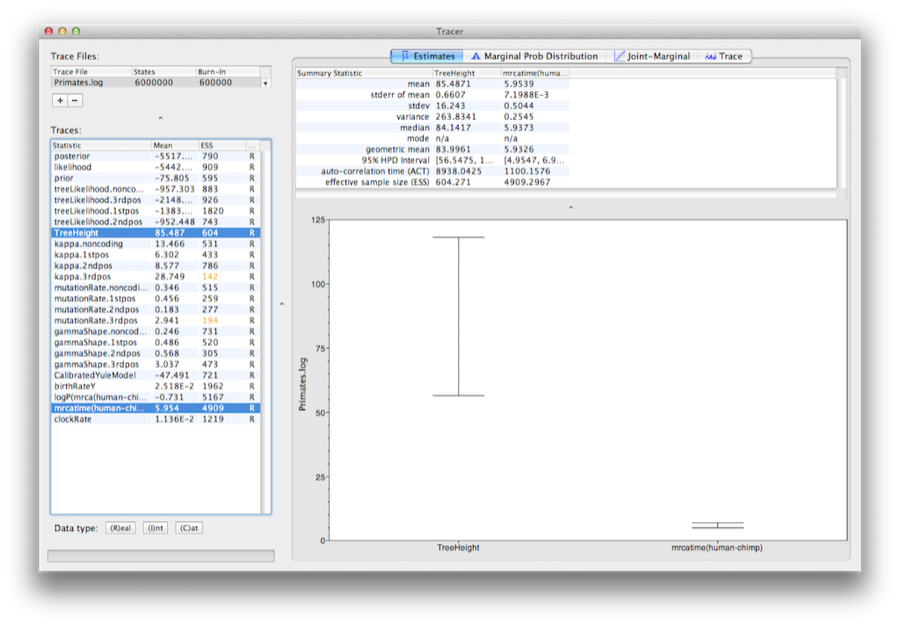
Figure 14: Uma captura de tela dos intervalos de 95% HPD da altura da raiz e do MRCA especificado pelo usuário (human-chimp) no Tracer.
5 Estimativas marginais posteriores
Para mostrar as taxas relativas para as quatro partições, selecione o parâmetro mutationRate para cada uma das quatro partições, e selecione a aba de densidade marginal no Tracer.A Figura 15 mostra as densidades marginais para as taxas relativas de substituição. O gráfico mostra que as posições 1 e 2 do códão têm taxas substancialmente diferentes (0,456versus 0,183) e ambas são muito mais lentas que a posição 3 do códão com uma taxa relativa de 2,941. A partição não codificadora tem uma taxa intermediária entre as posições do códão 1 e 2 (0,346). Em conjunto, este resultado sugere uma forte selecção purificadora tanto nas regiões codificadoras como não codificadoras do alinhamento.
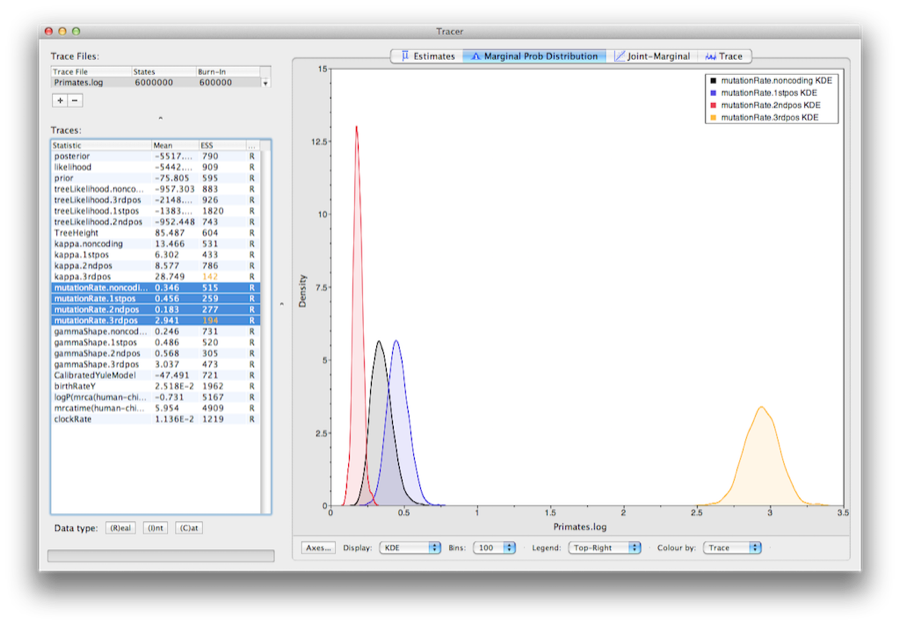
Figure 15: Uma captura de ecrã das densidades marginais posteriores das taxas relativas de substituição das quatro partições (em relação à taxa média ponderada pelo local).
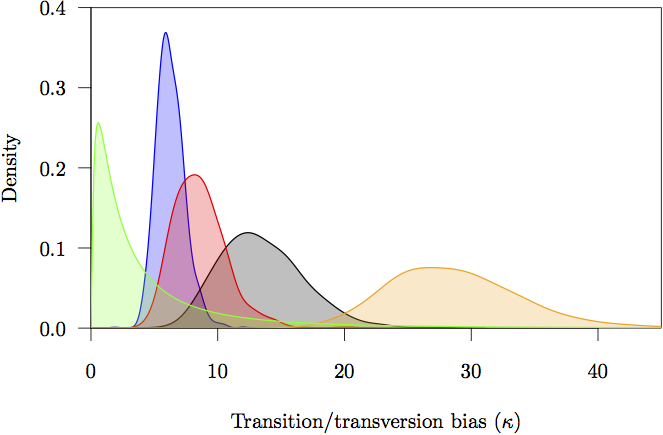
Figure 16: As densidades marginal anterior e posterior para os parâmetros de forma (α). O a priori está na cor cinza. A densidade posterior estimada para cada partição também é mostrada: sem codificação (laranja) e primeira (vermelho), segunda (verde) e terceira (azul) posições de códon.
>
Figure 17: As densidades marginal anterior e posterior para os parâmetros de viés de transição/tranversão (κ). O a priori está na cor cinza. A densidade posterior estimada para cada partição também é mostrada: sem codificação (laranja) e primeira (vermelho), segunda (verde) e terceira (azul) posições de códon.
Questões
Qual é a taxa estimada de evolução molecular para esta árvore genealógica (inclua o intervalo de 95% HPD)?
>
Que fontes de erro esta estimativa inclui?
Quantos anos tem a raiz da árvore (dê a média e o intervalo HPD 95%)?
6 Obtendo uma estimativa da árvore filogenética
BEAST também produz uma amostra posterior de árvores filogenéticas de tempo juntamente com a sua amostra de estimativas de parâmetros. Estas precisam ser resumidas usando o programa TreeAnnotator. Isto irá pegar o conjunto de árvores e encontrar a mais bem suportada. Ele então anotará esta árvore representativa de resumo com a média de idades de todos os nós e as faixas correspondentes de 95% de HPD. Ele também calculará a probabilidade de clade posterior para eachnode. Execute o programa TreeAnnotator e configure-o como mostrado na Figura 18.
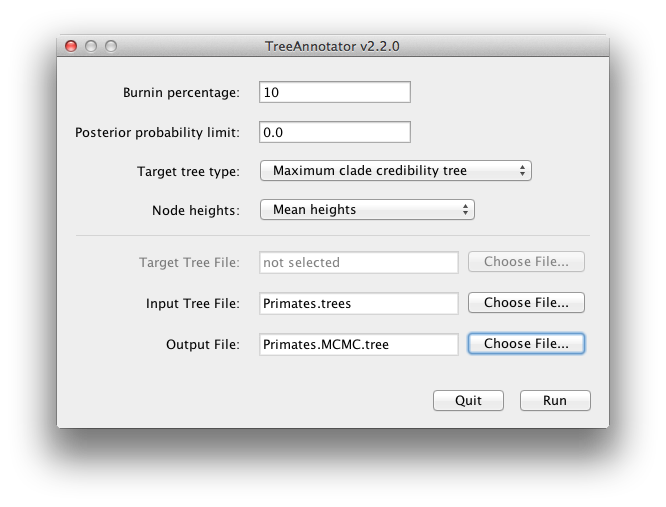
Figure 18: Uma captura de tela do TreeAnnotator.
The burnin é o número de árvores a serem removidas do início da amostra. Ao contrário do Tracer que especifica o número de passos como uma queimadura, no TreeAnnotator você precisa especificar o número real de árvores. Para esta execução, você especificou um comprimento de cadeia de 6.000.000 passos de amostragem a cada 1.000 passos. Assim, o arquivo de árvores conterá 6.000 árvores e assim para especificar uma queima de 10% no campo de texto superior.
A opção de limite de probabilidade Posterior especifica um limite tal que se um nó for encontrado a menos desta freqüência na amostra de árvores (ou seja, tem uma probabilidade posterior menor que este limite), ele não será anotado. O padrão de 0,5 significa que apenas nodesseen na maioria das árvores será anotado. Defina isto para zero para anotar todos os nós.
O tipo de árvore alvo especifica a topologia da árvore que será anotada. Você pode escolher uma árvore específica de um arquivo ou pedir ao TreeAnnotator para encontrar uma árvore na sua amostra. A opção padrão, árvore de máxima credibilidade de clade, encontra a árvore com o produto mais alto da probabilidade posterior de todos os seus nós.
Para alturas de nós, o padrão é Common Ancestor Heights, que calcula a altura de um nó como a média do tempo MRCA de todos os pares de nós na clade. Para árvores com grande incerteza na topologia e, portanto, muitas clades com baixo suporte, alguns outros métodos podem resultar em árvores com ramos de comprimento negativo. Nesta análise, o suporte para todos os clades na árvore resumida é muito alto, portanto isto não é problema aqui. Escolha Alturas médias para as alturas dos nós. Isso define as alturas (idades) de cada nó na árvore para a altura média através de uma amostra de árvores para aquela clade.
Para o arquivo de entrada, selecione o arquivo de árvores que BEAST criou e selecione um arquivo para a saída (aqui o chamamos de Primates.MCC.tree). Agora pressione Run e aguarde o programa terminar.
7 Visualizando a árvore estimativa
Finalmente, podemos visualizar a árvore em outro programa chamado FigTree. Execute este programa, e opte pelo arquivo Primates.MCC.tree usando o comando Open no menu File. A árvore deve aparecer. Agora você pode tentar selecionar algumas das opções no painel de controle do lado esquerdo. Em primeiro lugar, gaste a opção Trees no painel, e marque Order nodes e escolha Ordering por decrescimento. Tente selecionar Barras de Nó para obter as barras de erro da idade do nó. Também ative as Rótulos de Ramo e selecione posterior a getit para exibir a probabilidade posterior para cada nó. Se você usar um modelo de relógio não restrito então em Appearance você também pode dizer a FigTree para colorir os ramos pela taxa. Você deve terminar com algo similar a Figura 19.
>
>
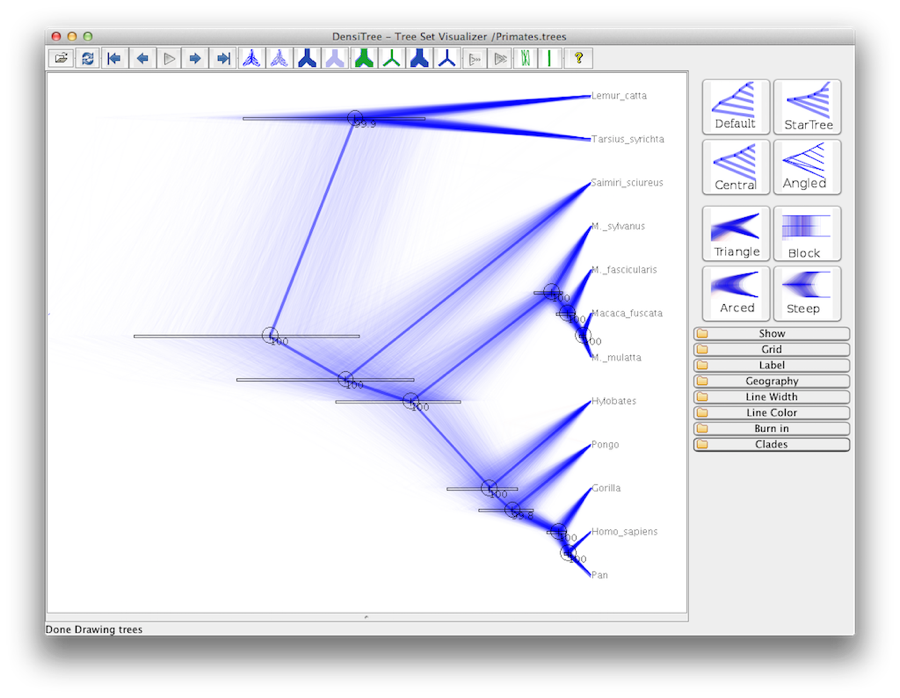
>Figure 19: Uma captura de tela da FigTree e DensiTree.
Uma visão alternativa da árvore pode ser feita com DensiTree, que é parte da Besta 2. A vantagem do DensiTree é que ele é capaz de visualizar tanto a incerteza nas alturas dos nós quanto a incerteza na topologia. Então, concluímos que esta análise resulta em um consenso muito alto sobre a topologia (Figura 19).
Questões
- A taxa de evolução difere substancialmente entre as diferentes linhagens da árvore?
- DensiTree tem uma barra de clades (Menu Window/View clade toolbar) para mostrar informações sobre clades.
Qual é o suporte para o clade ?
- Você pode navegar pelas topologias no DensiTree usando o menu Browse.A topologia mais popular tem um suporte de mais de 99%.
Qual é o suporte para a segunda topologia mais popular?
- Atrás do menu de ajuda, o DensiTree mostra algumas informações.
Quantas topologias estão no conjunto de árvores?
8 Comparando os seus resultados com os anteriores
É uma boa ideia repetir a análise enquanto se procede à amostragem do anterior para ter a certeza de que as interacções entre os antecedentes não estão a afectar as suas informações anteriores. Ainteração entre os priores pode ser problemática especialmente quando se usa calibração, pois significa colocar múltiplos priores na árvore.
Usando BEAUti, configure a mesma análise mas sob as opções MCMC, selecione a opção Amostra do anterior apenas. Isto irá permitir-lhe visualizar a distribuição prévia completa na ausência dos seus dados de sequência. Resumir as árvores a partir da distribuição prévia completa e comparar o resumo com a árvore de resumo posterior.
A estimativa do tempo de divergência usando “datação por nó” do tipo descrito neste capítulo foi aplicado para responder a uma variedade de diferentes questões em ecologia e evolução. Por exemplo, a datação por nós com fósseis foi usada para determinar a diversidade de espécies de cycadáceas, analisar a taxa de evolução em plantas floridas e investigar as origens das cianobactérias quentes e frias do deserto .
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond e Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), no. 1, 114. A Gelman, G Roberts, and W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled e Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138–49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, e S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian, e Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith e Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008),no. 5898, 86–89.
Este documento foi traduzido de LATEX byHEVEA.