John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Det finns allt fler bevis för det nära sambandet mellan diabetes typ II (T2D) och Alzheimers sjukdom (AD). Det är viktigt att dessa två sjukdomar har ett antal patologiska likheter, bland annat amyloidansamling, oxidativ stress, inflammation och celldöd. Hittills saknas läkemedelsterapier för Alzheimers sjukdom och T2D, och det finns ett stort behov av att upptäcka och utveckla nya terapier för dessa sjukdomar. Ett antal studier på människor och gnagare har visat att tillskott av metaboliska hormoner är mycket värdefullt för att förbättra den kognitiva funktionen och den allmänna metaboliska hälsan vid både T2D och Alzheimers sjukdom. Pankreashormonet amylin har framstått som en viktig komponent i sjukdomsetiologin för både T2D och AD, även om den exakta roll som amylin spelar i dessa sjukdomar ännu inte är väl förstådd. Här granskar vi kritiskt den aktuella litteraturen som använder humant amylin eller dess syntetiska analog, pramlintid, samt amylinreceptorantagonister för behandling av AD.
Introduktion
Alzheimers sjukdom (AD) är en progressiv, försvagande neurodegenerativ sjukdom som kännetecknas av ackumulering av amyloid-beta (Aβ)-plack och neurofibrillära trassel som består av hyperfosforylerat tau1. Ackumulationen av dessa patologiska peptider bidrar till brister i exekutiva funktioner som inlärning och minne, humör, affekt osv. och utgör en betydande börda för patienten och vårdgivarna. Förekomsten av Alzheimers sjukdom ökar i alarmerande takt i USA, med uppskattningsvis 5,5 miljoner amerikaner som lever med Alzheimers sjukdom 2017 och detta antal förväntas tredubblas fram till 20502. Dessutom överstiger kostnaderna för vård och behandling av AD-patienter för närvarande 200 miljarder dollar per år och förväntas bara öka3. Även om Alzheimers sjukdom helt klart är ett monumentalt problem i och utanför USA är behandlingsalternativen fortfarande mycket begränsade4. Många läkemedelsförsök har genomförts med ett stort antal målinriktade metoder, men det finns för närvarande endast sex läkemedel som godkänts av FDA för Alzheimers sjukdom och som endast är symtomatiska behandlingar5, 6. Hittills har majoriteten av de farmakologiska medel som utvecklats varit specifikt inriktade på Aβ- eller tau-patologin, men ingen av dem har lyckats avlägsna eller förebygga patologin4. Det finns därför ett grundläggande behov av att utveckla fungerande terapier och förebyggande behandlingar för Alzheimers sjukdom.
Åldersrelaterad (sporadisk) Alzheimers sjukdom är en komplicerad multifaktoriell sjukdom som har många genetiska och miljömässiga influenser. Miljö och livsstil är starkt involverade i utvecklingen av sporadisk Alzheimers sjukdom; faktorer som kost7-9, fetma8-10, metaboliskt syndrom7, typ II-diabetes (T2D)9, 11 och kardiovaskulära sjukdomar12 har alla involverats i orsakerna till Alzheimers sjukdom. Det är av avgörande betydelse att antalet fall av fetma och diabetes ökar snabbt parallellt med Alzheimers sjukdom12, 13. Även om sambandet mellan fetma och Alzheimers sjukdom är något oklart finns det bevis för att fetma mitt i livet spelar en roll för utvecklingen av Alzheimers sjukdom10. Ännu viktigare är att fetma ofta åtföljs av ett antal andra sjukdomar, bland annat kardiovaskulära sjukdomar, högt blodtryck, dyslipidemi, T2D, stroke osv.14. Förekomsten av T2D ökar snabbt och CDC uppskattar att cirka 30,3 miljoner människor (1 av 10 vuxna) i USA har diabetes och att hela 84,1 miljoner (1 av 3 vuxna) har prediabetes, varav de flesta är omedvetna om sitt tillstånd. På grund av en storskalig minskning av fysisk aktivitet som åtföljs av en samtidig ökning av matintag och dålig kost föreslås dessutom att antalet fall av fetma, T2D, metaboliskt syndrom och kardiovaskulära sjukdomar bara kommer att öka till uppskattningsvis 600 miljoner fall av T2D i världen fram till 203515.
Det finns många bevis för att metabolisk funktion och sjukdom är inblandade i processen för kognitiv försämring och åldrande16, 17. Till exempel rapporterar cirka 70 % av de personer som fått diagnosen T2D kognitiv försämring och ett stort antal T2D-patienter utvecklar senare demens16, 18-21. Personer som diagnostiserats med T2D i minst fem år har en betydligt ökad risk att utveckla Alzheimers sjukdom jämfört med personer som har lidit av T2D i mindre än fem år17. Tillsammans tyder dessa uppgifter på att den ökande förekomsten av T2D i befolkningen kan bidra till de ökande frekvenserna av AD.
T2D kännetecknas initialt av högt blodglukos och insulin, vilket leder till hyperinsulinemi; viktigt är att amylin, ett litet metaboliskt hormon som produceras av β-islet-cellerna i bukspottkörteln, sampaketeras och samutsöndras tillsammans med insulin och därmed överproduceras vid T2D22. Viktigt är att det finns ett antal patologiska kännetecken som finns i både T2D och AD: 1) minskad metabolism i hjärnan och metabolisk hormonresistens 2) amyloidpatologi 3) oxidativ stress (OS) och inflammation. Kronisk hyperinsulinemi och hyperamylinemi leder till ett antal fysiologiska problem: kronisk hyperinsulinemi leder till insulinresistens i systemet22, försämrad insulintransport över blod-hjärnbarriären (BBB)23, 24 och därmed minskad insulinsignalering i hjärnan25. Förlust av insulinsignalering i hjärnan är förknippad med ett antal AD-relaterade patologiska särdrag, inklusive ökad Aβ-produktion, tau-fosforylering och neuroinflammation.
För övrigt har amylin liknande patologiska särdrag som Aβ vid höga koncentrationer26 och kan vara en gemensam väg mellan de två sjukdomarna. Exempelvis har amylinfibriller hittats i bukspottkörteln hos 95 % av T2D-patienterna27-29 och orsakar ett antal fysiologiska störningar, inklusive avvikande Ca2+-inflöde, ökad sekretion av proinflammatoriska cytokiner30,31 och i slutändan förlust av β-isletceller32. Dessutom passerar amylin lätt BBB och bildar amylinfibriller samt blandade plack med Aβ i hjärnan och kan vara ansvarig för AD-liknande patologi och Aβ-utsöndring vid T2D33-35. Amylin är känt för att påverka långtidspotentiering (LTP) i hippocampus och kan ha ett inneboende inflytande på den kognitiva funktionen i hjärnan36-39. Det är dock fortfarande oklart om amylin är en toxisk insult i dessa sjukdomar eller om dess funktionella förlust genom aggregering eller förlust av β-celler i sent skede vid T2D bidrar till utvecklingen av en AD.
The Amylin Signaling Dichotomy
Det råder fortfarande stor debatt om inblandningen av amylinreceptorn (AMYR) och amylinsignalering i sjukdomsförloppet och etiologin vid T2D och AD. Forskningen som syftar till att urskilja detta förhållande expanderar snabbt. All relevant forskning har konsekvent visat att modulering av amylinsignalering påverkar AD-relaterad patologi. Hur detta förhållande ser ut har dock ännu inte klarlagts på ett konkret sätt. Flera grupper har producerat övertygande data som tyder på att amylinsignalering är fördelaktigt när det gäller att förebygga AD-relaterad patologi och kognitiva brister både in vivo och in vitro40-44. Det är viktigt att pramlintid, en rekombinant icke-aggregerande form av amylin, som används tillsammans med insulinbehandlingar för att behandla diabetes och som förbättrar den glykemiska kontrollen, minskar kroppsvikten och minskar serummarkörer för OS45-47, också är lovande som ett läkemedel mot Alzheimers sjukdom. Hittills har det dock inte gjorts några kliniska prövningar som syftar till att använda amylin eller pramlintid som ett terapeutiskt medel för behandling av demens. Tydliga bevis från gnagarstudier tyder på att kronisk behandling med antingen humant amylin eller pramlintid innebär starka terapeutiska fördelar när det gäller att minska AD-relaterad patologi; tillägg av amylin/pramlintid minskar nivåerna av lösligt Aβ, plackbörda, tau-fosforylering, neuroinflammation och OS samtidigt som kognitionen förbättras40-42,44. Ovanstående data tyder på att en förlust av medfödd amylinsignalering i CNS på grund av aggregering ger upphov till en ökad risk för utveckling av AD och behandlas mer i detalj i Grizzanti et al. 201848.
I kontrast till detta visar studier också att humant amylin och Aβ har liknande toxiska effekter och att dessa toxiska effekter kan lindras med hjälp av AMYR-antagonist36-39,49. Data visar till exempel att behandling in vivo med AMYR-antagonister ger mycket liknande fysiologiska fördelar som behandling med amylin eller pramlintid. Behandling av TgCRND8 AD-möss med AC253, en AMYR-antagonist, eller dess cykliska motsvarighet cAC253 minskar neuroinflammation, nivåer av lösligt Aβ och plackbörda samtidigt som kognitionen förbättras50. På samma sätt visar in vitro/ex-vivo-studier att låga doser humant amylin eller Aβ orsakar störningar i LTP och att dessa brister blockeras av AC253 eller pramlintid38,39. Högre doser humant amylin/amylinoligomerer är förknippade med okontrollerat Ca2+-inflöde, vilket är starkt kopplat till celldöd26,32. Tillsammans stöder dessa uppgifter en toxisk funktion hos amylinoligomerer och därmed en potentiell terapeutisk mekanism för AMYR-blockad. Däremot har andra visat att de positiva effekterna av amylin kan blockeras med hjälp av AC25341. Den terapeutiska potentialen för behandling eller hämning av amylin förblir således oklar och belyser den komplexa och dikotoma karaktären hos amyloider i hjärnan och periferin.
Pussel ihop pusslet
Det finns ett antal hål i den nuvarande litteraturen som behöver fyllas för att ge en mer komplett bild av amylinhistorien: 1) naturen av det medfödda amylinsystemet och amylinsignalering i hjärnan 2) Aβ- och pramlintid-signaleringens förmåga genom de tre viktigaste AMYR-receptorerna och relaterade receptorer 3) de terapeutiska mekanismer genom vilka amylin/pramlintid- eller AMYR-hämning förmedlas. För det första visar intressanta nya data att AMYR inte bara är involverad i signalering utan även i ligandtransport över BBB. AMYR är en heterodimär receptor som består av en calcitoninreceptor och ett receptoraktivitetsmodifierande protein (1-3)51 . En 50-procentig global knockdown av calcitoninreceptorn (en nyckelkomponent i AMYR) minskade signifikant den mängd AC253 som hittades i hjärnan50 , vilket tyder på att AMYR som är lokaliserade i BBB är involverade i transporten av dessa ligander in i hjärnan och kan också vara involverade i att transportera amylin och pramlintid in i/ut ur hjärnan. Förekomsten av dessa transportmekanismer i BBB tyder på att amylin sannolikt har en inneboende fysiologisk funktion i hjärnan, eftersom dess transport in i hjärnan är hårt kontrollerad. Hur amylinsignalering eller brist på amylin leder till de patologiska egenskaperna hos Alzheimers sjukdom och om AMYR är det medel genom vilket Aβ förmedlar sina toxiska effekter är dock fortfarande oklart.
Nästan finns det motstridiga bevis när det gäller förhållandet mellan Aβ och AMYR. Även om flera studier tydligt visar att humant amylin och Aβ har liknande effekter på LTP i CNS och användning av AMYR-hämmare förbättrar dessa skadliga effekter36-39, tyder andra bevis på att Aβ (1-42) är oförmögen att signalera genom någon av AMYR för att framkalla någon form av cAMP-svar vid en mängd olika koncentrationer52. Det är möjligt att Aβ aktiverar olika signalkaskader genom interaktion med AMYR eller helt enkelt fungerar som en inert kompetitiv hämmare, men detta har ännu inte påvisats.
För övrigt visade en separat studie att oligomeriskt amylin förmedlar sina toxiska effekter direkt genom AMYR och indirekt genom TRPV4, en icke-selektiv katjonkanal26. Låga koncentrationer av humant amylin framkallar ett Ca2+-svar som förmedlas via dess ursprungliga receptor. Vid högre koncentrationer bildar humant amylin dock oligomerer och aktiverar avvikande signalering som resulterar i aktivering av TRVP4-kanaler och möjliggör okontrollerat katjoninflöde, särskilt Ca2+. Farmakologisk blockering av AMYR och TRPV4 visar att båda receptorerna är nödvändiga för att oligomeriskt humant amylin ska kunna framkalla sina toxiska Ca2+-effekter26. Det är därför troligt att Aβ förmedlar sina toxiska effekter på AMYR på ett liknande sätt, även om det ännu inte finns några uppgifter om detta. Okontrollerat Ca2+-inflöde är kopplat till ett antal patologiska fenomen, inklusive okontrollerad vesikulär frisättning, OS- och mitokondriell dysfunktion, apoptos osv. Därför är det troligt att cellulär dysfunktion och utvecklingen av ytterligare AD-liknande patologi som uppstår till följd av toxisk amyloidsignalering förmedlas genom både AMYR och TRPV4. Det är därför nödvändigt att urskilja de signalkaskader som modulerar förhållandet mellan AMYR och TRVP4. Vidare är farmakologiska experiment motiverade som använder Aβ och pramlintid i ett brett spektrum av doser för att fastställa Aβ och pramlintids effekter på Ca2+-strömmar, LTP, cAMP-produktion och andra signalkaskader för att fastställa deras signaleringsförmåga. Dessa experiment kommer att bidra till att fylla några av tomrummen i den nuvarande litteraturen när det gäller AMYR och dess inblandning i sjukdomstillstånd (figur 1).
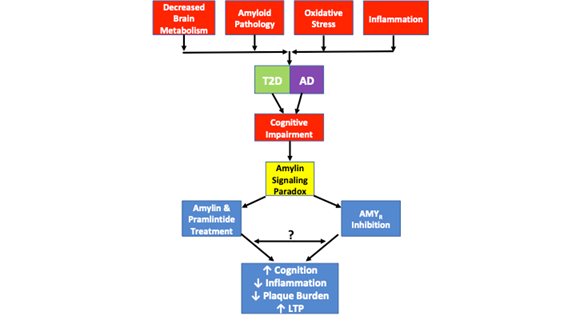
Figur 1. visar amylinsignaleringsparadoxen och patologiska likheter som observerats i T2D och AD. Minskad metabolism i hjärnan, amyloidpatologi, oxidativ stress och inflammation är alla gemensamma patologiska drag som observeras i båda sjukdomarna. Även om inte alla fall av T2D eller AD innehåller var och en av dessa patologiska egenskaper, uppvisar alla fall kognitiv försämring. Amylinsignaleringsparadoxen kommer in i bilden eftersom studier har visat att både AMYR-hämning och AMYR-agonism via behandling med amylin och pramlintid resulterar i förbättrad kognition, minskad inflammation, minskad plackbörda och ökad LTP. De signalmekanismer som styrs av AMYR-agonism och AMYR-antagonism har ännu inte helt klarlagts. Medan amylinsignalering traditionellt förknippas med cAMP- och PKA-signalering är det oklart om andra kaskader också aktiveras av amylin/pramlintid. Vidare är det oklart om AMYR-antagonister, amylinoligomerer eller Aβ signalerar genom AMYR eller om det finns några likheter eller överlappningar mellan alla dessa AMYR-ligander. Som sådan kommer ett antal experiment som föreslås i denna översikt att bidra till att ytterligare belysa AMYR:s verkliga natur.
Slutsatser
Den nuvarande olikheten när det gäller amylinsignaleringens roll i hjärnan visar att det finns ett väsentligt behov av att ytterligare belysa amylins inblandning i både Alzheimers sjukdom och T2D. Vid T2D är det troligt att amylin i de tidiga stadierna av sjukdomen översvämmas av hjärnan, bildar oligomerer, inducerar avvikande signalering genom sin ursprungliga receptor och rekryterar TRPV4 för att inducera patologiskt Ca2+-inflöde som resulterar i utbredd neuronal dysfunktion som yttrar sig i form av OS, okontrollerad vesikulär frisättning och interneuronal dysfunktion, inflammation och resulterande celldöd. Denna mekanism kan vara ansvarig för den inledande övergången från den friska hjärnan till hjärnans åldrande vid metabola sjukdomar. Som sådan kan AMYR- eller TRVP4-hämning vid vissa tidpunkter i metabolisk sjukdom och i de tidiga stadierna av diabetes vara motiverad för att blockera de toxiska effekterna av oligomeriskt amylin eller Aβ. Starka bevis tyder dock också på att amylinersättning med antingen humant amylin eller pramlintid minskar de flesta av de viktigaste AD-relaterade patologierna samtidigt som kognitionen förbättras i gnagarmodeller av AD. Därför kan det vara motiverat att ersätta amylinsignalering med amylin eller pramlintid i de mellersta till sena stadierna av diabetes när amylinsignalering går förlorad på grund av aggregering, oligomerisering eller förlust av β-celler. I detta syfte finns det också ett behov av att urskilja den tidsmässiga presentationen av patologiska händelser vid metaboliskt kopplat hjärnåldrande och terapeutiska alternativ för tidiga, mellanliggande och sena sjukdomsstadier. Kritisk analys och testning av dessa amyloiders direkta natur och signalfunktioner samt den terapeutiska karaktären hos specifika temporala behandlingar kan bidra till att överbrygga klyftan mellan AMYR-hämningsbehandlingar och amylinersättningsbehandlingar.
Finansiering
Finansiering för denna artikel tillhandahölls av National Institutes of Aging grant 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau och synaptisk dysfunktion. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013; 80: 1778-1783.
- Association As. 2016 Alzheimer’s disease facts and figures. Alzheimers & Demens. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glukosnivåer och risk för demens. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of dementia prevalence in the United States and China. Fetma. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Typ 2-diabetes som riskfaktor för Alzheimers sjukdom: störande faktorer, interaktioner och neuropatologi i samband med detta förhållande. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Fetma och dess komorbida tillstånd. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF diabetes atlas. Bryssel: Internationella diabetesfederationen. 2013.
- K Dash S. Cognitive impairment and diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk för demens bland personer med diabetes mellitus: en befolkningsbaserad kohortstudie. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. The lancet Diabetes & endokrinologi. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus och Alzheimers sjukdom: gemensam patologi och behandling. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalens av diabetes och prediabetes och deras riskfaktorer bland vuxna i Bangladesh: en landsomfattande undersökning. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insulinnedbrytande enzym reglerar nivåerna av insulin, amyloid β-protein och den intracellulära domänen av β-amyloid prekursorprotein in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulinbindning till hjärnans kapillärer minskar hos genetiskt överviktiga, hyperinsulinemiska Zuckerråttor. Peptider. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Insulinnivåerna minskar i cerebrospinalvätskan hos kvinnor med prodomal Alzheimers sjukdom. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neuropharmakologi. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mekanismer som kopplar fetma till insulinresistens och typ 2-diabetes. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Försämrad glukostolerans är förknippad med ökad islet amyloid polypeptide (IAPP) immunoreaktivitet i betaceller i bukspottkörteln. The American Journal of Pathology. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatoriska markörer och risk för typ 2-diabetes: en systematisk genomgång och metaanalys. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimers β-amyloid, humant islet amylin och prionproteinfragment framkallar intracellulära fria kalciumförhöjningar genom en gemensam mekanism i en hypotalamisk GnRH-neuronal cellinje. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal amylinavlagring, peroxidativ membranskada och ökad IL-1β-syntes i hjärnor från Alzheimers sjukdomspatienter med typ 2-diabetes och i diabetiska HIP-råttor. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo sådd och korsning av lokaliserad amyloidos: en molekylär länk mellan typ 2-diabetes och Alzheimers sjukdom. Den amerikanska tidskriften för patologi. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylinreceptorn: ett gemensamt patofysiologiskt mål vid Alzheimers sjukdom och diabetes mellitus. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ)-peptid aktiverar direkt amylin-3-receptorsubtypen genom att utlösa flera intracellulära signalvägar. J Biol Chem. 2012; 287: Pramlintide Antagonizes Beta Amyloid (Aβ)-and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloid-inducerad depression av hippocampal långtidspotentiering medieras genom amylinreceptorn. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Intraperitoneal injektion av bukspottkörtelpeptiden amylin minskar kraftigt beteendestörningar och amyloidpatologi i hjärnan i murinmodeller av Alzheimers sjukdom. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylinreceptorligander minskar den patologiska kaskaden av Alzheimers sjukdom. Neuropharmacology. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotektiva effekter av amylinanalogen pramlintide på Alzheimers sjukdomspatogenes och kognition. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilisering av β-catenin genom mutationer i presenilin-1 potentierar neuronal apoptos. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylinbehandling minskar neuroinflammation och förbättrar onormala mönster av genuttryck i hjärnbarken i en musmodell för Alzheimers sjukdom. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetate injection for the treatment of type 1 and type 2 diabetes mellitus. Clin Ther. 2007; 29: 535-562.
- Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta?analysis. Diabetes, fetma och ämnesomsättning. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Action of β-amyloid protein on human neurons are expressed through the amylin receptor. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyklisk AC253, en ny amylinreceptorantagonist, förbättrar kognitiva brister i en musmodell av Alzheimers sjukdom. Alzheimers & Demens: Translational Research & Clinical Interventions. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reducerat nociceptivt beteende i islet amyloid polypeptid (amylin) knockout-möss. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Aktivitet av pramlintid, amylin från råtta och människa men inte Aβ1-42 vid mänskliga amylinreceptorer. Endokrinologi. 2014; 155: 21-26.