Introduktion
Germline de novo mutationer (DNMs) är genetiska förändringar hos individen som orsakas av mutagenes som sker i föräldrarnas könsceller under oogenes och spermatogenes. Här ska termen ”de novo” inte förväxlas med termen ”ny mutation”. Trots att DNM i samband med en trio (far, mor och barn) är nya mutationer kan de vara vanliga, sällsynta eller nya varianter i den allmänna befolkningen. För att mäta och förklara frekvensen av en viss DNM är det nödvändigt att först bedöma variantens inverkan på fenotypen, eftersom nya gynnsamma egenskaper kan utvecklas när uppkomna genetiska mutationer erbjuder en specifik överlevnadsfördel (Front Line Genomics, 2017).
I människor med genetiska icke-mendelska sjukdomar som uppträder sporadiskt är DNM:er vanligen nya, mer tillförlitliga och skadligare än nedärvda varianter, eftersom de inte utsätts för starkt naturligt urval (Crow, 2000; Front Line Genomics, 2017). Att identifiera den genetiska orsaken till en sjukdom som induceras av en DNM hos en individ kan därför vara en utmaning ur klinisk synvinkel, eftersom pleiotropi och genetisk heterogenitet kan ligga till grund för en enda fenotyp (Eyre-Walker och Keightley, 2007). Följaktligen har man under det senaste decenniet gjort stora ansträngningar för att sekvensera exomer från individer med sjukdomar med oklar genetisk etiologi i syfte att ställa klinisk diagnostik. Även vid upptäckt av kandidatvarianter de novo finns det dock fortfarande otillräcklig information om de vanliga och sällsynta varianterna, vilket utesluter en tydlig slutsats om den identifierade de novo-variantens patogenicitet och dess roll i sjukdomen (Acuna-Hidalgo et al., 2016). Denna begränsning kan förklaras av att de novo-varianter vanligtvis är heterozygota och kan vara antingen extremt sällsynta eller vanliga. I fall av mycket sällsynta de novo-varianter kan variantens patogenicitet vara svår att bevisa eftersom det inte finns fler patienter med samma fenotyp och de novo-variant. När det gäller vanliga de novo-varianter är det möjligt att man inte vet vilka faktorer som bestämmer manifestationerna av variantens patogenicitet, särskilt om vissa individer i den allmänna befolkningen har varianten men inte har den genetiska sjukdomen. Oavsett frekvensen av de novo-varianter kan dock båda typerna av varianter skalas på grundval av relativ lämplighet och naturligt urval.
Anpassningsbarheten beror på många faktorer; för att bedöma om en DNM är patogen eller adaptiv, och för att förstå varför den förekommer med en viss frekvens i populationen, är det därför nödvändigt att undersöka varianten under lämpliga förhållanden. Dessa inkluderar miljö, föräldrarnas ålder, genomisk kontext, epigenetik och andra faktorer eftersom alla dessa faktorer påverkar värdet av den genomsnittliga relativa fitnessen som ökar monotont, medan styrkan i urvalet minskar (Peck och Waxman, 2018).
Det huvudsakliga syftet med den här studien var att belysa frekvensen av förekommande DNM:er och bestämma hur dessa mutationer är fördelade i exomerna i den allmänna litauiska befolkningen. Vi undersökte också om frekvensen av dessa mutationer påverkades av sammansättningen eller strukturella parametrar i de sekvenser där de förekom och andra faktorer som skulle kunna påverka de mekanismer som ligger till grund för bildandet av dessa DNMs. Slutligen försökte vi fastställa om DNM:erna uppstod på grund av ett intensivt tryck från det naturliga urvalet på de funktionella regionerna. Även om fördelningen och intensiteten av DNMs har varit föremål för många studier hade de inte tidigare undersökts i den litauiska befolkningen.
Material och metoder
I den här studien analyserade vi prover från den litauiska befolkningen som erhållits från LITGEN-projektet (LITGEN, 2011). Datamängden bestod av 49 trios med totalt 144 olika individer. Genomiskt DNA extraherades från venöst blod med hjälp av antingen fenolklorform-extraktionsmetoden eller den automatiserade DNA-extraktionsplattformen TECAN Freedom EVO® (Tecan Schweiz AG, Schweiz) baserad på den paramagnetiska partikelmetoden. Exomer sekvenserades på ett SOLiD 5500-sekvenseringssystem (75 bp reads). Sekvenseringsdata bearbetades och förbereddes med Lifescope-programvaran. Exomerna kartlades i enlighet med det mänskliga referensgenomet byggd 19. Det genomsnittliga läsdjupet för sekvensering var 38,5. BAM-formaterade filer för mor, far och barn som genererades av Lifescope kombinerades med hjälp av SAMtools-programvaran för varje trio.
De novo-mutationer identifierades med hjälp av två programvaror: VarScan (Koboldt et al., 2012) och VarSeqTM. En potentiell variant ansågs vara en DNM om den identifierades i avkomman men inte fanns i någon av föräldrarna på samma position. Totalt sett upptäcktes 1 752 och 4 756 DNM med VarScan respektive VarSeqTM. För att avfärda falskt positiva de novo-anrop, när det var okänt om alla individer i trion identifierades korrekt, tillämpades konservativa filter på detekterade DNM-kvalitetsparametrar enligt följande: (1) individens genotypkvalitet ≥50, (2) antal läsningar på varje plats >20. SnpSift-programvaran användes för att tillämpa dessa filter på de data som genererades av VarScan. Data som genererades av VarSeqTM -programvaran filtrerades genom att välja samma filtreringsparametrar i segmentet Trio Workflow. För att dessutom förkasta de återstående varianterna som var somatiska (endast närvarande i en bråkdel av de sekvenserade blodcellerna) med låg allelbalans eller sekvenseringsartefakter filtrerades DNM:er genom att ställa in ett tröskelvärde för den observerade bråkdelen av läsningarna hos individer med den alternativa allelen (allelbalansen) för trion (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Dessutom granskades alla möjliga identifierade och filtrerade de novo singelnukleotidvarianter manuellt med Integrative Genomics Viewer (Robinson et al., 2011). På grund av det stora antalet identifierade DNM:er valdes 51 de novo enskilda nukleotidvarianter slumpmässigt ut för validering av varianter genom Sanger-sekvensering. Sangersekvensering utfördes med hjälp av en ABI PRISM 3130xl Genetic Analyzer. Alla filtrerade och manuellt granskade DNM som identifierats med VarScan (N = 95) och VarSeqTM (N = 84) annoterades med hjälp av ANNOVAR (Butkiewicz och Bush, 2016; Wang et al., 2010). För analys av proteininteraktioner användes programvaran STRING (Szklarczyk et al., 2017). Liksom i fallet med exomkartläggning utfördes annoteringarna med hjälp av hg19 referensgenom för människa.
Sannolikheten för att en anropande position var en DNM i trion beräknades oberoende av varandra för varje trion. Som beskrivits i en tidigare referens (Besenbacher et al, 2015) beräknades de novo-frekvensen per position per generation (PPPG) enligt följande:
där f är antalet trion och N är antalet anropsbara positioner, som potentiellt kan identifieras som de novo-positioner för varje trio separat, oavsett sekvenseringsdjup. Detta antal varierar beroende på trio. ni är antalet identifierade DNM-platser för trio i. Sannolikheten Pji (de novos ingle nucleotide) för att den kallade enskilda nukleotidplatsen j och familjen i ska vara muterad beräknades enligt följande:
Sannolikheten Pji (de novo indel)för att den kallade indelplatsen j och familj i skall muteras beräknades enligt följande:
där C, M och F står för avkomma, mor respektive far och Hetero, HomR och HomA står för heterozygot, homozygot för referens och homozygot för alternativ allel. Sannolikheten Pij (de novo) beräknades med hänsyn till sekvenseringstäckningen. Konfidensintervall för skattningar av hastigheten beräknades på samma sätt som för binomiala proportioner. För uppskattning av DNM-frekvensen och för ytterligare beräkningar använde vi R-paketet (version 3.4.3) (R Core Team, 2013).
För att testa hypotesen att variationer i DNM-frekvensen i olika regioner av genomet kan förklaras av inneboende egenskaper hos själva genomregionen och föräldrarnas ålder utfördes en linjär regressionsanalys, för vilken den ”sekundära” annotationen av varje DNM utfördes med hjälp av data från projekten ENCODE (ENCODE Project Consortium, 2012) och LITGEN (LITGEN, 2011). Först valdes enligt en tidigare studie (Besenbacher et al., 2015), för att samla in uppgifter om det genomiska landskapet för de identifierade DNM:erna, lymfoblastoida cellinjer (LCL och GM12878) (ENCODE Project Consortium, 2012) för att samla in uppgifter om det genomiska landskapet för de identifierade DNM:erna. Data samlades in för:
(1) uttrycksfrekvenser (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) i olika vävnader. Enligt uttryck av regioner med DNMs delades in i positioner med specifikt och icke-specifikt uttryck;
(2) mätningar av DNase1 överkänslighetsställen (DHS). DHS-status tilldelades 0 om den var utanför DHS-toppen och 1 om den var inom;
(3) mätningar av kontexten av CpG-öar. Om DNM låg inom CpG-öar tilldelades status för position 1; om utanför – 0;
(4) tre histonmärken (H3K27ac, H3K4me1 och H3K4me3) från ENCODE-projektet. Om DNM befann sig i en position markerad med histon tilldelades den 1 och om inte – 0;
(5) GERPP++-bevaringsvärden samlades in med hjälp av annoteringsverktyget ANNOVAR. Enligt bevarandevärdena tilldelades positioner med DNM i konservativa (GERP++-poäng >12) och icke-konservativa positioner (GERP++-poäng <12) (Davydov et al., 2010; ENCODE Project Consortium, 2012). Baserat på frågeformulärsposter från LITGEN-projektet samlades uppgifter om föräldrarnas ålder in. Efter insamling av parametrar för varje trio beräknades ett antal positioner med varje parameter. Därefter utfördes en korrelationsanalys följt av linjär regressionsmodellering av DNM-frekvens och parametrar.
Resultat
Efter DNM-analysen identifierades ett exceptionellt högt antal DNM för två trion (nr 4 och 21): 113 och 123 (med VarScan respektive VarSeqTM) och 16 (VarScan). Dessa fynd föranledde oss att testa biologiskt faderskap, vilket förkastades för trio nr. 4 och bekräftades för trio nr. 21. Uppgifterna för trio nr. 4 inte med i studien. I den slutliga uppsättningen av 48 trios identifierades 95 DNM i 34 trios med VarScan-programvaran och 84 DNM i 31 trios identifierades med VarSeqTM-programvaran (figur 1). Ingen DNM upptäcktes i 18 och 15 trios med VarScan respektive VarSeqTM. Av alla DNM:er som identifierades av båda programvarorna matchade endast 5,37 % av DNM:erna (tre DNM:er i MEIS2-, PGK1- och MT1B-gener). Varje person hade i genomsnitt 1,9 (VarScan-programvaran) och 1,7 (VarSeqTM) DNMs.
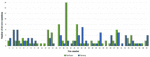
FIGUR 1. Jämförelse av de novo singelnukleotidvarianter som identifierats av VarScan (blå) och VarSeqTM (grön) programvara.
Analys av 95 DNM som identifierades av VarScan programvara visade att 20 DNM var exoniska, inklusive två stop-gain DNM, sju synonyma DNM och 11 icke-synonyma DNM. Åttio nya mutationer som identifierades med VarSeqTM var exoniska, inklusive 1 stop-gain DNM och 78 icke-synonyma DNM (figur 2). Majoriteten av de DNM som identifierades av VarScan fanns i kromosomerna 1, 2, 4 och 5, medan VarSeqTM identifierade DNM främst i kromosomerna 2, 6, 7 och 11. Antalet identifierade DNM:er korrelerade inte med tätheten av gener i kromosomerna (R = 0,09, p-värde = 0,65 för VarScan och R = 6,73, p-värde = 0,51 för VarSeqTM) eller med kromosomstorleken (figur 3). Enligt de båda programvarorna var förhållandet mellan övergångar och transversioner mycket lika: 1,44 respektive 1,47 (figur 4). Skillnader i strukturerna för övergångarna identifierades dock. Närmare bestämt fanns det bland DNM som identifierades av VarScan fler G/T- och A/C-övergångar, medan det bland DNM som identifierades av VarSeqTM fanns fler A/T- och G/C-övergångar.
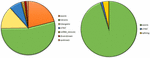
FIGUR 2. Sammansättningen av de novo mutationer (DNM) som genererats av VarScan (till vänster) och VarSeqTM (till höger).
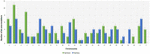
FIGUR 3. Fördelning av antalet de novo-varianter per kromosom enligt VarScan och VarSeqTM genererade data. Gröna staplar representerar DNMs som identifierats av VarScan-programvaran, blå – av VarSeqTM. Felstaplarna representerar standardfelet för medelvärdet av DNMs för varje kromosom.
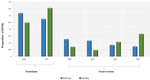
FIGUR 4. De molekylära händelser som ligger till grund för övergångar inträffar oftare än de som leder till transversioner, vilket resulterar i en ∼1,5-faldigt högre frekvens av övergångar än transversioner över hela exomet. Övergångs- och transversionshändelser identifierade med VarScan (grönt) och VarSeqTM (blått) programvaran. Felstaplarna representerar standardfelet för de genomsnittliga DNMs.
De beräknade frekvenserna av de novo enskilda nukleotidsmutationer var 2,4 × 10-8 PPPG (95 % konfidensintervall : 1,96 × 10-8-2,99 × 10-8) enligt VarSeqTM och 2.74 × 10-8per nukleotid per generation (95 % CI: 2,24 × 10-8-3,35 × 10-8) enligt VarScan.
Tre de novo indels i tre trios identifierades av VarScan-algoritmen i kromosomerna 6 och 11. Den beräknade andelen de novo indels i genomet var 1,77 × 10-8 (95 % CI: 6,03 × 10-9-5,2 × 10-8) PPPG. Det är anmärkningsvärt att alla de novo indels var ”reversibla”, dvs. föräldrarna hade nya varianter i genomet och deras barn hade de novo-varianter baserade på referensgenomet med 37,5 medelvärde för sekvenseringsdjupet respektive 50 genotypkvalitet. Dessa tre DNM valdes dock inte ut för validering med hjälp av Sangersekvenseringsmetoden, vilket innebär att sannolikheten för en överskattning av de novo indels ändå kvarstår. De novo indels var C/T och A/G i samband med enskilda nukleotider.
Linjär regressionsmodellering visade att DNAse 1-överkänslighetsställen, kontext av CpG-öar, GERPP++-konserveringsvärden och uttrycksnivåer förklarade ∼68-93 % av DNM-frekvensen (tabell 1). Varken epigenetiska markörer eller faderns ålder korrelerade signifikant med DNM-frekvensen. Modellerna upprättades endast utifrån data från VarScan eftersom det inte fanns någon korrelation mellan data från VarSeqTM och inneboende egenskaper hos själva genomregionen.

TABELL 1. Den linjära regressionen av DNAaseI-överkänslighetsställen, kontext av CpG-öar, GERPP++-konserveringsvärden och uttrycksnivåens effekt av på hastigheten av DNMs.
Funktionell förutsägelse av DNMs
För att bedöma vilka missense-mutationer som var skadliga och förändrade funktionen hos det drabbade proteinet efter typ, analyserades förutsagda kategoriska poäng för den skada som induceras av DNMs. Följande 10 värden beaktades: polyphen HDIV och HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, Fathmm-MKL-kodning och GERP++. På grundval av de förutspådda värdena valdes fyra DNM:er ut som identifierades av VarScan som hade sex eller fler skadliga eller troligen skadliga förutsägelser. Dessa stop-gain DNM:er fanns i generna MEIS2 och ULK4, medan icke-synonyma DNM:er fanns i generna MT1B och PGK1. Proteiner som kodas av dessa gener är viktiga för neuronal tillväxt, endocytos och skydd mot de negativa effekterna av tungmetaller. Dessa proteiner deltar i frisättningen av tumörblodkärlshämmaren angiostatin och i olika signalvägar. Det fanns inga samband mellan de proteiner som kodas av dessa gener (figur 5).
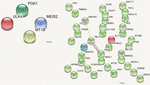
FIGUR 5. Protein-proteininteraktioner (Szklarczyk et al., 2017) i gener som hyser DNMs. DNMs identifierade med VarScan i gener som kodar proteiner är till vänster, DNMs identifierade med VarSeqTM – till höger. Färgade linjer indikerar ett samband mellan proteiner.
De novo-mutationer som identifierats av VarSeqTM analyserades mer i detalj om de förutspåddes vara skadliga eller troligen skadliga av minst hälften av prediktionsverktygen. Det fanns 35 punktmutationer (se ??) i gener som kodar för proteiner som var viktiga för kromatinremodellering, reglering av cytoskelettet, celltillväxt och livskraft, cytoplasmatiska signalvägar och initiering av neuronala reaktioner som utlöser luktuppfattning.
Av de proteiner som kodades av de DNM-affekterade generna var endast CLPTM1, ZNF547 och DMXL1 sammankopplade på något sätt (figur 5).
Diskussion
I den här studien genomförde vi en omfattande analys av fördelningen av DNM:er över olika regioner av exomet i den litauiska befolkningen. Totalt upptäcktes 95 DNMs i 34 trios och 84 DNMs i 31 trios med hjälp av sekvenseringstekniken SOLiD 5500 genom algoritmerna VarScan respektive VarSeqTM. Först och främst vill vi notera att vi valde VarScan för identifiering av DNMs eftersom denna algoritm enligt (Warden et al., 2014) resulterar i en lista över varianter med hög överensstämmelse (>97 %) med högkvalitativa varianter som identifierats av GATK UnifiedGenotyper och HaplotypeCaller. VarSeqTM-programvaran valdes eftersom den är ett allmänt använt verktyg för variantanalys både inom forskning och klinisk analys. Trots att båda algoritmerna är utformade för att söka efter DNM i avkommans exom som inte fanns hos någon av föräldrarna, var överensstämmelsen mellan de två programvarorna för DNM-analys endast 5,37 %. VarScan-algoritmen hade högre känslighet (5,42 %) för DNM-detektering före filtrering än VarSeqTM-algoritmen (1,77 %), varför vi misstänkte att inget verktyg lyckades kalla mutationer på grund av hög känslighet som alltid åtföljdes av låg specificitet. Därför föreslår vi att en avsevärd förbättring av resultaten kan uppnås genom att kombinera resultatet från olika verktyg (Sandmann et al., 2017).
Baserat på de genererade uppgifterna var den uppskattade DNM-frekvensen för enskilda nukleotider mellan 2,4 × 10-8 och 2,74 × 10-8 och den för de novo indels var 1,77 × 10-8 PPPG, beroende på vilken algoritm som användes. Vår beräknade DNM-frekvens var högre än den som rapporterats i tidigare studier (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015), där den varierade mellan 1,2 × 10-8 och 1,5 × 10-8 PPPG. Den högre DNM-frekvensen i vår studie var rimlig eftersom vår studie baserades på exomdata. Dessutom uppvisar exomer betydligt högre (med 30 %) mutationsfrekvens än hela genomer eftersom basparsammansättningen i hela genomet skiljer sig från den i exomer. Framför allt har exomer ett genomsnittligt GC-innehåll på cirka 50 %, medan det för hela genomet är cirka 40 % (Neale et al., 2012). Metylerade CpGs utgör mycket mutabla sekvenser hos människor på grund av den spontana deamineringen av cytosinbaser (Neale et al., 2012). Enligt jämförande genomikstudier tros den ökade mutationsfrekvensen vid CpG-rika regioner ha utvecklats kring tiden för däggdjurens strålning (Francioli et al., 2015). Under arternas divergens genomgick CpG-rika exoniska regioner ökade mutationshastigheter jämfört med dem på icke-kodande DNA och förvandlades till icke-kodande regioner. Därför, då effekten av CpG-innehållet minskar med tiden, minskar den genomsnittliga mutationshastigheten tills den når den nivå som finns i det omgivande icke-kodande DNA (Subramanian och Kumar, 2003). Medan sekvenser i neutralt evolverande regioner av genomet har haft tillräckligt med tid att jämna ut sig med avseende på dinukleotidkontexter, har dock renodlande urval upprätthållit hypermutabla CpGs i funktionella regioner (Subramanian och Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Eftersom vi fann en högre DNM-frekvens än den som rapporterats i andra studier spekulerade vi därför att den åtminstone delvis kan bero på den lokala sekvenskontexten och/eller ett eventuellt naturligt urvalstryck på exomet. Följaktligen tillämpades en linjär regressionsmodell, och vi fann att DNAse 1-överkänslighet, kontext av CpG-öar, GERPP++-konserveringsvärden och uttrycksnivå förklarade ∼68-93 % av DNM-frekvensen. Dessa resultat visade att DNM:er i exomet bildades oberoende av bevarandet av DNA-sekvenser. DNM-frekvensen var dock högre i gener vars produkter var ospecifika och i transkriptionellt aktiva promotorliknande regioner.
I motsats till resultaten från andra studier (Wong et al., 2016; Sandmann et al., 2017) fann vi att faderns ålder inte korrelerade med DNM-frekvensen. Dessa resultat kan förklaras av att datamängden bestod av trios med liknande föräldraålder och att endast en liten del (∼1,5 %) av hela genomet analyserades. Baserat på dessa parametrar hade varje person i genomsnitt endast 1,9 (VarScan) eller 1,7 (VarSeqTM) DNM jämfört med 40-82 i hela genomet (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al, 2016), medan antalet de novo indels i den kodande sekvensen liknade det som identifierades i (Front Line Genomics, 2017).
Resultaten av vår omfattande funktionella analys av annotationer avslöjade att av alla identifierade DNM:er var 4 (VarScan) och 35 (VarSeqTM) varianter sannolikt patogena DNM:er. Skillnaden i antalet patogena DNMs kan förklaras av att beroende på vilken algoritm som används för identifiering av DNMs, skiljer sig andelen DNMs i kodande sekvenser avsevärt. Till exempel var 21,05 % av de DNM som identifierades med VarScan-programvaran exoniska, medan 95,24 % av de som identifierades med VarSeqTM-programvaran var exoniska. Dessa patogena DNM fanns i gener som kodar för proteiner som är viktiga för kromatinmodellering, reglering av cytoskelettet, modulering av celltillväxt och vitalitet, funktion av cytoplasmatiska signalvägar och initiering av neuronala reaktioner. Trots att dessa DNM anses vara patogena identifierade sig alla personer som deltog i undersökningen som genetiskt ”friska”. Därför tyder detta resultat på att trots DNM:s förmodade patogenicitet tolererade de genom i vilka DNM:s fanns uppenbarligen sådana förändringar, så att sjukdomsmanifestationer ofta inte var uttalade. Enligt Szamecz et al. (2014) gäller att ju oftare DNM:er förekommer i bevarade genetiska positioner, desto starkare är effekterna av det naturliga urvalet på genetiska förändringar genom kompensatoriska mekanismer för skydd av genomet. De skadliga effekterna av varianterna kan mildras på fyra sätt. Vissa gener kan tolerera trunkerade varianter av proteiner eftersom deras funktionella effekter maskeras av ofullständigt uttryck, kompenserande varianter eller låg funktionell betydelse av trunkeringen (Bartha et al., 2015). Däremot kompenseras genförändringar i samband med icke-synonyma DNM:er genom mekanismen för nyttig mutationsackumulering i hela genomet (Szamecz et al., 2014). Det tyder på att i dessa fall är de patogena mutationerna inte tillräckligt skadliga för att minska den genomsnittliga fitnessen och därför kvarstår de längre i många generationer och formas av det naturliga urvalet.
Sammanfattningsvis gav vår analys av fördelningen av DNM:er och deras genetiska och epigenetiska kontext insikter om den genetiska variationen i det litauiska genomet. Baserat på dessa resultat kan ytterligare studier i patientgrupper med genetiska sjukdomar underlätta vår förmåga att särskilja vissa patogena DNMs från de tolererade bakgrunds-DNMs och att identifiera tillförlitliga orsakande DNMs. Den främsta begränsningen i denna studie var dock att vi inte undersökte variationen i icke-kodande och regulatoriska genregioner. Denna information skulle kunna bidra till att belysa möjliga mekanismer för DNM-bildning som fortfarande inte är tillräckligt tydliga.
Accessionskoder
Sekvensdata har deponerats i European Nucleotide Archive (ENA), under accession PRJEB25864 (ERP107829).
Etikutlåtande
Denna studie genomfördes i enlighet med rekommendationerna för tillstånd, Vilnius Regional Ethics Committee for Biomedical Research. Protokollet godkändes av Vilnius regionala etikkommitté för biomedicinsk forskning. Alla försökspersoner gav skriftligt informerat samtycke i enlighet med Helsingforsdeklarationen.
Författarbidrag
LP utförde dataanalysen och förberedde manuskriptet. AJ beräknade andelen de novo mutationer. Sekvensering av trios exomer utfördes av LA och IK. VK var huvudforskare.
Finansiering
Denna studie stöddes av Europeiska socialfonden inom ramen för Global Grant-åtgärden. LITGEN-projekt nr. VP1-3.1-ŠMM-07-K-01-01-013.
Intressekonfliktförklaring
Författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller finansiella relationer som skulle kunna tolkas som en potentiell intressekonflikt.
Supplementärt material
Supplementärt material för denna artikel finns online på: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). Nya insikter om generering och roll av de novo-mutationer vid hälsa och sjukdom. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). Egenskaperna hos heterozygota proteintrunkerande varianter i det mänskliga genomet. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Ny variation och de novo-mutationsfrekvens i befolkningsomfattande de novo-sammansatta danska trios. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). Kluster av CpG-öar och proepigenetiskt urval för CpGs i proteinkodande exoner av HOX och andra transkriptionsfaktorer. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). In silico funktionell annotering av genomisk variation. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Ursprung, mönster och konsekvenser av människans spontana mutationer. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A. och Batzoglou, S. (2010). Identifiering av en stor del av det mänskliga genomet som är under selektiv begränsning med hjälp av GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Ett integrerat uppslagsverk över DNA-element i det mänskliga genomet. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., and Keightley, P. D. (2007). Fördelningen av fitnesseffekter av nya mutationer. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genombredda mönster och egenskaper hos de novo-mutationer hos människor. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics Magazine Issue 14 – ASHG. London: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Genetiska effekter på genuttryck i olika mänskliga vävnader. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: Upptäckt av somatiska mutationer och kopianummerförändringar i cancer genom exomsekvensering. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Frekvens av de novo mutationer och betydelsen av faderns ålder för sjukdomsrisken. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Finskaliga skillnader i rekombinationshastighet mellan kön, populationer och individer. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ’t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Transkriptom- och genomsekvensering avslöjar funktionell variation hos människor. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Tillgänglig på: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Mönster och frekvens av exoniska de novo-mutationer vid autismspektrumstörningar. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Vad är anpassning och hur ska den mätas? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). Ett språk och en miljö för statistiska beräkningar. Wien: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Utvärdering av verktyg för variant calling för icke-matchade Next-Generation Sequencing Data. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S. och Sunyaev, S. (2008). Hypermutabla icke-synonyma platser är under starkare negativt urval. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Neutrala substitutioner förekommer snabbare i exoner än i icke-kodande DNA i primatgenom. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). Det genomiska landskapet för kompensatorisk evolution Be. Det genomiska landskapet för kompensatorisk evolution. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). STRING-databasen 2017: kvalitetskontrollerade protein-proteinassocieringsnätverk som gjorts allmänt tillgängliga. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M. och Hakonarson, H. (2010). ANNOVAR: funktionell annotering av genetiska varianter från nästa generations sekvenseringsdata. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L. och Wu, X. (2014). Detaljerad jämförelse av två populära variant calling-paket för exom- och riktade exonstudier. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Nya observationer om moderns ålderseffekt på germina de novo mutationer. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar