Incidensen och prevalensen av njurstenssjukdom fortsätter att öka i befolkningen. Med en livstidsprevalens på upp till 10 % är nefrolithiasis (NL) och nefrokalcinos (NC) därför stora hälsobelastningar, särskilt i västvärlden (1). NL och NC är förknippade med betydande sjuklighet och utveckling till kronisk njursjukdom på grund av återfall, upprepade kirurgiska/endoskopiska ingrepp och samtidig inflammation. På en förenklad nivå är njurstensbildning resultatet av en obalans mellan urinhämmare (t.ex. citrat, magnesium, uromodulin och pyrofosfat) och främjare (t.ex. oxalat, kalcium, fosfat, urat och cystin) av kristallisering, som överskrider övermättnad med konsekutiv aggregering, kärnbildning och stentillväxt vid Randalls plack (figur 1). Denna obalans kan bero på förändrad enteral och/eller renal hantering av antingen främjare eller hämmare, såsom enteral malsekretion av oxalat eller renal malreabsorption av kalcium (figur 1).
Figur 1. Obalans mellan urinhämmare och främjare av kristallisering som leder till njurstensbildning. Koncentrationen av urinhämmare och promotorer påverkas och kontrolleras av både intestinala och renala transportörer (GI-njuraxeln). Dessa transportörer och utbytare, till exempel SLC26A1, är ansvariga för sekretion och absorption. När det gäller oxalat leder enteral hyperabsorption men malsekretion och/eller renal hypersekretion men malabsorption till att oxalatnivåerna i urinen överskrider övermättnaden och därmed främjar kristallisering och konsekutiv stenbildning .
Den underliggande etiologin till NL tros vara multifaktoriell med en miljömässig, notabel kost-, hormonell och genetisk komponent. I tvillingstudier har arvbarheten för njursten uppskattats till 56 % (3), och upp till två tredjedelar av hyperkalciuriska stenbildare har släktingar med NL (4). Även om kalciumhaltiga njurstenar står för mer än 80 % av alla, är den genetiska grunden för sådana stenar fortfarande till stor del okänd (5). Med undantag för varianter i CLDN14, TRPV5, SLC34A1, ALPL, CASR och UMOD har genomomövergripande associationsstudier ännu inte gett några betydande genetiska faktorer (6-8). Riskalleler har dock identifierats inom gener som också visat sig överföra sjukdomen på mendelsk grund, t.ex. CASR, SLC34A1 och SLC2A9 (9, 10). Hittills har mer än 30 enskilda gener med en Online Mendelian Inheritance in Man-definierad fenotyp identifierats som involverade i NL/NC, om de är muterade (tabell 1).

Tabell 1. Gener som är kända för att orsaka monogena former av NL/NC.
Förmåner för nedärvning av monogena former inkluderar autosomal-dominant, autosomal-recessiv och X-bunden överföring. Intressant nog har man för flera av dessa gener rapporterat både recessiva och dominanta arvsformer: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 och SLC4A1. Medan de flesta syndromiska och allvarliga medfödda sjukdomar uppvisar ett recessivt arvsmönster (Bartter, Lowe, Dent, FHHNC och distal renal tubulär acidos med sensorineural dövhet), är mildare tillstånd snarare förknippade med mutationer i dominanta gener. Majoriteten av de kodade proteinerna utgör renala lösörestransportörer (t.ex. SLC34A1, SLC34A3 och SLC9A3R1), men även kloridkanaler (CLCN5), tight-junction-proteiner (t.ex. CLDN16/CLDN19) och metaboliserande enzymer (t.ex. AGXT, APRT och CYP24A1) har visat sig vara defekta hos patienter med NL/NC. Därför är den underliggande defekten oftast belägen i själva njurens tubulära system och kan därför tillskrivas som tubulopati. Omvänt är a priori extrarenala tillstånd, som vid primär hyperoxaluri (PH) där dysfunktion av leverenzymer (AGXT, GRHPR och HOGA1) orsakar oxalatansamling med sekundär njurpåverkan, tänkbara orsaker till NL/NC. Även om varje sjukdomsfenotyp anses representera en relativt sällsynt enhet kan enstaka genorsaker stå för ett betydande antal patienter på grund av deras breda genetiska heterogenitet (42). Förutom genetisk heterogenitet finns det också en allelisk variation, där trunktionsvarianter snarare resulterar i funktionsförlust och missensevarianter (hypomorfa) kan orsaka ganska subtila defekter, som kan överblickas kliniskt, särskilt hos vuxna stenbildare. Ett annat nyligen uppskattat fenomen handlar om gendoseringseffekter i flera av de tidigare nämnda njurstensgenerna. I SLC34A3 till exempel, som kodar för en av de viktigaste fosfattransportörerna i den proximala tubuli (NaPiIIc), har det visats att heterozygota individer inte längre bara kan betraktas som friska bärare, eftersom de uppvisar njurförkalkningar och/eller benmanifestationer betydligt oftare än vildtypindivider, men fortfarande i mindre utsträckning än bialleliska (homozygota och sammansatta heterozygota) individer (43). Liknande observationer rapporterades för familjer med mutationer i CYP24A1 (44). Bidraget från monogena sjukdomar till den totala prevalensen av njurstenssjukdom har tidigare inte studerats på ett omfattande sätt. Framför allt saknas genetiska bevis baserade på breda screeningar av en mängd orsakande gener i stora patientkohorter. Omfattande genetiska tester har tidigare varit alltför kostsamma och ineffektiva. För de flesta individer med NL/NC har därför mutationsanalys för en orsakande genetisk defekt inte varit tillgänglig, trots att kunskap om den molekylära orsaken till NL/NC kan få viktiga konsekvenser för prognos, profylax och/eller behandling. Endast grova uppskattningar har kunnat härledas från kliniska observationsstudier: på grundval av en enorm datainsamling om analys av stensammansättning drogs slutsatsen att monogena orsaker inte överstiger 9,6 % hos barn och 1,6 % hos vuxna (45). Under det senaste decenniet har dock denna situation börjat förändras i och med tillkomsten av sekvenseringstekniker med hög genomströmning.
Höggenomströmningsmutationsanalys hos patienter med NL/NC
För att undersöka patienter med njurstenssjukdom med avseende på förekomst av patogena mutationer i kända sjukdomsgener har vi upprättat en genpanel som bygger på mikrofluidisk multiplex-PCR och konsekutiv NextGen-sekvensering (Fluidigm™/NGS) (46, 47).
I en ”pilotstudie” rekryterade vi efter varandra 268 genetiskt olösta individer från typiska njurstenskliniker; 102 pediatriska och 166 vuxna probands. Som ett resultat identifierade vi 50 skadliga varianter i 14 av 30 analyserade gener, vilket ledde till en molekylär diagnos i 15 % av alla fall. I den pediatriska undergruppen upptäckte vi en orsakande mutation i 21 %, medan det bland vuxna fanns skadliga varianter i 11 % (figur 2A) (48). Mutationer i cystinuriagenen SLC7A9 hittades mest frekvent i den vuxna kohorten (figur 2B). Två uppföljningsstudier kunde bekräfta dessa resultat. För det första förklarades 17 % av fallen i en uteslutande pediatrisk kohort med 143 NL/NC-patienter av mutationer i 14 olika gener (49). För det andra, i en kohort av 51 familjer med ålder för NL/NC-manifestation före 25 år, användes riktad WES för att upptäcka en genetisk orsak i nästan 30 % (50). Föga förvånande hittades recessiva mutationer oftare bland nyfödda barn och i fall av medfödd sjukdom, medan dominanta tillstånd vanligtvis manifesterades senare i livet. Dessa uppgifter tyder på att genetisk njurstenssjukdom är ett underdiagnostiserat tillstånd, trots att den molekylära diagnosen potentiellt kommer att påverka prognos, profylax och/eller behandling. En begränsning värd att nämna är dock en potentiell selektionsbias på grund av rekryteringen från specialiserade njurstenskliniker i alla de tre ovan nämnda studierna.

Figur 2. Mutationsanalys av 30 kända monogena gener för nefrolithiasis (NL)/nefrokalcinos (NC) hos 268 patienter med NL/NC. (A) Fraktion av monogena orsaker i den pediatriska och vuxna subkohorten. (B) Antal monogena orsaker i alla gener (rött anger vuxna, orange anger pediatriska patienter). Noterbart är att SLC7A9 hittades mest frekvent muterad, särskilt hos unga vuxna.
Identifiering av nya mänskliga sjukdomsgener med hjälp av kandidatgenmetoden
Mutationsanalyser med hög genomströmning används också för att söka efter patogena varianter i olika kandidatgener. Ett av de mest intressanta fynden på senare tid var upptäckten av mänskliga mutationer i SLC26A1 (32). Sedan den första beskrivningen av Ca-oxalat (CaOx) njurstensbildning och NC hos Slc26a1 (Sat1)-knockoutmöss av Dawson et al. 2010 har SLC26A1 varit en bona fide NL-kandidatgen (51). SLC26A1 kodar för en anjonbytare som uttrycks vid det basolaterala membranet i proximala njurtubuli, ileum och jejunum. Genom att använda en kandidatgenmetod identifierades följaktligen patogena varianter hos människor med en historia av tidig CaOx-NL, nämligen två obesläktade individer med bialleliska missense-varianter (32). Funktionellt visades patogenitet hos de identifierade varianterna in vitro, vilket ledde till intracellulär feltransport och försämrad transportaktivitet (32). Defekt SLC26A1 utgör därför en ny orsak till CaOx-NL och bör beaktas när man testar individer för orsaker till återkommande CaOx-stenbildning.
Nytt kliniskt patientregister för ärftlig njurstenssjukdom
De flesta epidemiologiska data om ökande prevalens i västvärlden härrör från amerikanska databaser. Även om det finns ett brådskande behov finns det för närvarande inga centraliserade europeiska databaser. Eftersom tidigare nämnda genetiska studier om prevalensen av ärftlig njurstenssjukdom utfördes med små kohorter från specialiserade centra i både Europa och USA, är en översättning till den allmänna situationen i Europa inte giltig. Medan Rare Kidney Stone Consortium i USA utgör en plattform som integrerar och samordnar registerverksamhet, grundforskning och klinisk forskning om sällsynta tillstånd som cystinuri, PH, APRT-brist, Dent- och Lowe-sjukdom, har ingen jämförbar datainsamling om patienter med ärftlig njurstenssjukdom genomförts vare sig i Europa eller i Tyskland i dag. I samarbete med det befintliga europeiska PH-registret OxalEurope (professor Bernd Hoppe, universitetet i Bonn) och genom finansiering från Deutsche Forschungsgemeinschaft och Else Kröner-Fresenius Stiftung inrättade vi nyligen ett kliniskt patientregister för ärftlig njurstenssjukdom vid universitetet i Leipzig. Registret får nationellt stöd av de tyska sällskapen för nefrologi för vuxna (DGfN) och barnnefrologi (GPN). Det är dessutom registrerat i det tyska registret över kliniska prövningar (DRKS-ID: DRKS00012891). Som en grundläggande del av studiens rekrytering erbjuds mutationsanalyser med hög kapacitet för kända och nya njurstensgener på forskningsbasis för patienter utan fastställd molekylär diagnos men med en klinisk bild som pekar på en underliggande genetisk mottaglighet: t.ex. tidig debutålder (<40 år), positiv familjehistoria, indikativa fenotyper som NC, cystinuri eller RTA, och allvarligt recidiverande NL (>3×) (tabell 2). Medan patienter med en redan fastställd genetisk diagnos i allmänhet tas med, inkluderas inte fall med sekundära NL/NC-orsaker, t.ex. malignitet, sarkoidos och primär hyperparatyreoidism, i den genetiska analysen. För att aktivt rekrytera patienter behöver ett kliniskt centrum vanligtvis ett godkännande från den lokala institutionella granskningsnämnden; en process för vilken vi erbjuder vår hjälp och assistans genom att tillhandahålla respektive mallar. Efter etiskt godkännande kan samtyckesblankett och kliniska datablad (t.ex. kliniskt frågeformulär) laddas ner från vår webbplats för registret (http://www.mks-registry.net). För att säkerställa en grundlig klinisk fenotypning kommer vi att be om omfattande patientinformation, t.ex. etnicitet, släktskap, familjehistoria, debutålder, recidiv (definierat som varje förmodad ny njurstenshändelse), dagligt vätskeintag, kirurgiska ingrepp och extrarenal inblandning med mera. Dokumenten kan fyllas i av patienten med hjälp av den inskrivande läkaren. Dessutom kommer biokemiska serumparametrar, inklusive kreatinin, eGFR, PTH, D-vitamin, elektrolyter, urinsyra och urinanalys (pH, kalcium, fosfat, magnesium, urinsyra, citrat, oxalat och cystin, företrädesvis från 24-timmarsurin, om inte punkturin), samt uppgifter om analys av stensammansättningen att begäras vid inskrivningen. Med hänsyn till att 24-timmars urinsamling och analys av stensammansättning inte utförs rutinmässigt vid alla institutioner, inkluderar vi dessa parametrar när de är tillgängliga. Kliniska uppföljningsbesök vartannat år av de inskrivna patienterna är önskvärda men inte obligatoriska. Efter registrering kommer rekryterande kliniska centra att få en personlig inloggning för att lägga in patientdata via vår webbplats för registret (http://www.mks-registry.net). Alternativt erbjuder vi inmatning av data elektroniskt när de skickas till oss i pappersform. De inmatade uppgifterna kommer att lagras på en säker server och kan nås av deltagande kliniska centra för att se sina egna patientuppgifter. På följande webbplatser finns ytterligare information:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
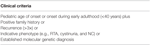
Tabell 2. Inklusionskriterier för mutationsanalys i kliniska patientregister.
Sammanfattningsvis är njurstenssjukdom ett allt vanligare tillstånd som är kliniskt heterogent och dåligt förstått, särskilt dess genetiska drivkrafter. Som en rad nyligen genomförda studier visade, är monogena tillstånd sannolikt underskattade i prevalens. Genom att införa ett centraliserat patientregister över ärftliga njurstenssjukdomar kommer vi att bidra till att åtminstone delvis överbrygga den stora kunskapsluckan när det gäller genetik för njurstenssjukdomar. I detta sammanhang är kliniska register värdefulla källor av flera skäl: för det första kommer det att vara viktigt att fastställa bättre samband mellan fenotyp och genotyp för att kunna stratifiera patienterna på ett mer exakt sätt i framtida kliniska forskningsstudier. För det andra kommer identifiering av nya sjukdomsgener med nya sjukdomsmekanismer att minska luckan med okänd NL/NC-etiologi, och för det tredje hjälper dechiffrering av nya molekylära mål till att bana väg för utveckling av läkemedel för förebyggande av återfall i svårt drabbade familjer.
Etikutlåtande
Denna studie genomfördes i enlighet med rekommendationerna från ”Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” med skriftligt informerat samtycke från alla försökspersoner. Alla försökspersoner gav skriftligt informerat samtycke i enlighet med Helsingforsdeklarationen. Protokollet godkändes av ”Ethikkommission an der Medizinischen Fakultät der Universität Leipzig.”
Författarnas bidrag
JH utformade och skrev manuskriptet. BH, AS, LM och RS redigerade manuskriptet och byggde upp registrets infrastruktur som presenteras för läsaren.
Intressekonflikter
Författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller ekonomiska relationer som skulle kunna tolkas som en potentiell intressekonflikt.
Finansiering
Forskningen finansieras av projektbidrag från DFG (HA 6908/2-1) och EKFS (2016_A52) till JH. Detta arbete stöddes dessutom av det federala ministeriet för utbildning och forskning (BMBF), Tyskland, FKZ: 01EO1501 till JH.
1. Scales CD Jr, Smith AC, Hanley JM, Saigal CS. Prevalens av njursten i Förenta staterna. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Genetik för hyperkalciurisk nefrolithiasis: njurstenssjukdom. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Framsteg i förståelsen av genetiken för kalciuminnehållande nefrolithiasis. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576.
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sekvensvarianter i CLDN14-genen associeras med njursten och benmineraltäthet. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones- role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Vanliga och sällsynta varianter som är associerade med njursten och biokemiska egenskaper. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 påverkar urinsyrakoncentrationerna med uttalade könsspecifika effekter. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutationer i glukostransportör 9 genen SLC2A9 orsakar renal hypouricemi. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identifiering och karakterisering av en gen med bassubstitutioner som är förknippade med den absorptiva hyperkalciuriefenotypen och låg spinal bentäthet. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Felriktad inriktning av peroxisomalt L-alanin:glyoxylataminotransferas till mitokondrier hos patienter med primär hyperoxaluri beror på aktivering av en kryptisk mitokondriell inriktningssekvens genom en punktmutation. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Mänskligt adeninfosforibosyltransferas. Identifiering av alleliska mutationer på nukleotidnivå som orsak till fullständig brist på enzymet. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutationer i ATP6N1B, som kodar för en ny vakuolär protonpumpsunderenhet 116-kD i njurarna, orsakar recessiv distal renal tubulär acidos med bevarad hörsel. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutationer i genen som kodar för B1-underenhet av H+-ATPas orsakar renal tubulär acidos med sensorineural dövhet. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II deficiency syndrome in a Belgian family is caused by a point mutation at an invariant histidine residue (107 His→Tyr): complete structure of the normal human CA II gene. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. Ett familjärt syndrom av hypokalcemi med hyperkalciuri på grund av mutationer i kalciumsensorreceptorn. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. En gemensam molekylär grund för tre ärftliga njurstenssjukdomar. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutationer i kloridkanalgenen CLCNKB orsakar Bartters syndrom typ III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, ett renalt tight junction-protein som krävs för paracellulär Mg2+-resorption. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutationer i tight-junction-genen claudin 19 (CLDN19) är förknippade med renal magnesiumförlust, njursvikt och allvarlig ögonpåverkan. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutationer i CYP24A1 och idiopatisk infantiel hyperkalcemi. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nefrokalcinos (emaljrenalsyndrom) orsakad av autosomalt recessiva FAM20A-mutationer. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Genen som kodar för hydroxypyruvatreduktas (GRHPR) är muterad hos patienter med primär hyperoxaluri typ II. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. HNF4A R76W-mutationen orsakar atypiskt dominant Fanconis syndrom utöver en β-cellfenotyp. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutationer i DHDPSL är ansvariga för primär hyperoxaluri typ III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identifiering av 17 oberoende mutationer som är ansvariga för brist på humant hypoxantin-guanin fosforibosyltransferas (HPRT). Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetisk heterogenitet hos Bartters syndrom avslöjad genom mutationer i K+-kanalen ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartters syndrom och MAGED2-mutationer. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Tightly linked flanking markers for the Lowe oculocerebrorenal syndrome, with application to carrier assessment. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartters syndrom, hypokalaemisk alkalos med hyperkalciuri, orsakas av mutationer i Na-K-2Cl-kotransporter NKCC2. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutationer i SLC26A1 orsakar nefrolithiasis. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molekylär identifiering av en renal uratanjonbytare som reglerar bloduratnivåerna. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditär hypofosfatrikos med hyperkalciuri orsakas av mutationer i genen SLC34A3 för natriumfosfatkotransporter. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinuri orsakad av mutationer i rBAT, en gen som är involverad i transport av cystin. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familjär distal renal tubulär acidos är förknippad med mutationer i genen för anjonbytare i röda blodkroppar (Band 3, AE1). J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Cystinuri som inte är av typ I orsakad av mutationer i SLC7A9, som kodar för en underenhet (bo,+AT) av rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1-mutationer och respons på renalt bisköldkörtelhormon. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Suggestiva bevis för en känslighetsgen nära D-vitaminreceptorlocus vid idiopatisk kalciumstensbildning. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identifiering av två mutationer i den humana xanthindehydrogenasgenen som är ansvariga för klassisk xanthinuri av typ I. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Hildebrandt F. Genetiska njursjukdomar. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutationer i SLC34A3/NPT2c är associerade med njursten och nefrokalcinos. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Kliniska och biokemiska fenotyper hos vuxna med monoalleliska och bialleliska CYP24A1-mutationer: bevis för gen-doseffekt. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Nya rön för diagnostik och behandling. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. Mutationsanalys med hög kapacitet hos patienter med nephronophthisis-associerad ciliopati med tillämpning av multiplexerad barcoded array-baserad PCR-amplifiering och nästa generations sekvensering. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identifiering av 99 nya mutationer i en världsomspännande kohort av 1 056 patienter med en nefronoftisrelaterad ciliopati. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. 14 monogena gener står för 15 % av nefrolithiasis/nefrokalcinos. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalens av monogena orsaker hos pediatriska patienter med nefrolithiasis eller nefrokalcinos. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar