Alexei Drummond, Andrew Rambaut, Remco Bouckaert och Walter Xie
1 Introduktion
Denna handledning introducerar BEAST-programvaran för bayesiansk evolutionär analys genom en enkel handledning. Handledningen omfattar samskattning av en genfylogeni och tillhörande divergenstider i närvaro av kalibreringsinformation från fossila bevis.
Du behöver följande programvara till ditt förfogande:
- BEAST – det här paketet innehåller programmet BEAST, BEAUti, TreeAnnotator och andra hjälpprogram. Den här handledningen är skriven för BEAST v2.2.x, som har stöd för flera partitioner. Den kan laddas ner från http://www.beast2.org/.
- Tracer – detta program används för att utforska utdata från BEAST (och andra Bayesianska MCMC-program). Det sammanfattar grafiskt och kvantitativt fördelningarna av kontinuerliga parametrar och ger diagnostisk information. Vid skrivande stund är den aktuella versionen v1.6. Den kan laddas ner från
http://tree.bio.ed.ac.uk/software/. - FigTree – detta är ett program för att visa och skriva ut molekylära fylogenier, i synnerhet de som erhållits medBEAST. I skrivande stund är den aktuella versionen v1.4.2. Den kan laddas ner från http://tree.bio.ed.ac.uk/software/.
Denna handledning kommer att vägleda dig genom analysen av en anpassning av sekvenser från tolv primatarter (se figur 1). Målet är att uppskatta fylogenin, utvecklingshastigheten för varje linje och åldrarna för de okalibreradeancestrala divergenserna.
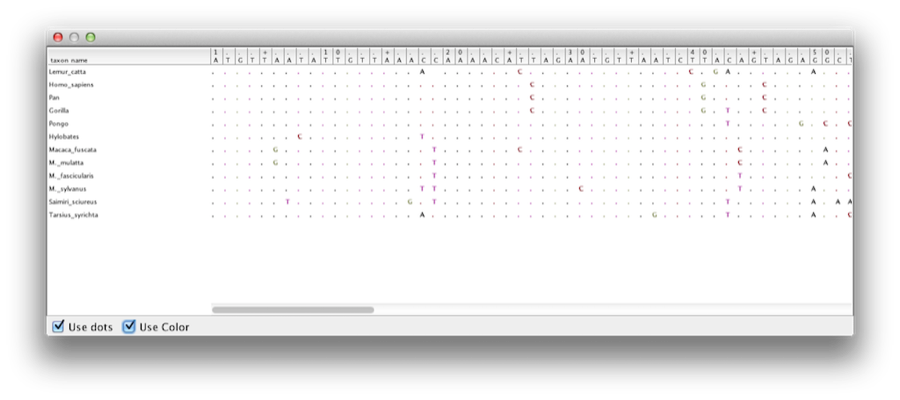
Figur 1: Del av anpassningen för primater.
Det första steget kommer att vara att konvertera en NEXUS-fil med ett DATA- eller CHARACTERS-block till en BEAST XML-inmatningsfil. Detta görs med hjälp av programmet BEAUti (som står för Bayesian Evolutionary Analysis Utility). Detta är ett användarvänligt program för att ställa in den evolutionära modellen och alternativen för MCMC-analysen. Det andra steget är att faktiskt köra BEAST med hjälp av den inmatningsfil som genereras av BEAUTi och som innehåller data, modell och analysinställningar. Det sista steget är att utforska BEAST:s resultat för att diagnostisera problem och sammanfatta resultaten.
2 BEAUti
Programmet BEAUti är ett användarvänligt program för att ställa in modellparametrarna för BEAST. Kör BEAUti genom att dubbelklicka på dess ikon. När BEAUti väl är igång kommer det att se likadant ut oavsett vilket datorsystem det körs på. I denna handledning används Mac OS X-versionen i figurerna, men Linux- och Windows-versionerna kommer att ha samma layout och funktionalitet.
2.1 Laddning av NEXUS-filen
För att ladda in en justering i NEXUS-format väljer du helt enkelt alternativet Importera justering… från menyn Filer, eller drar filen in i mitten av panelen Partitioner.
Exempelfilen med namnet primate-mtDNA.nex finns tillgänglig i katalogen examples/nexus/ för Mac och Linux och examples/nexus/ för Windows i katalogen där BEAST installerades.Filen innehåller en anpassning av sekvenser från 12 arter av primater.
Ett fönster Add Partition (Figur 2) öppnas om det relaterade paketet är installerat. Om du använder ”rent” BEAST 2 kan du gå till nästa stycke. I annat fall väljer du Add Alignment och klickar på OK för att fortsätta.
Figur 2: Fönstret Add Partition (visas endast om relaterade paket är installerade).
Om det finns några kodningsöverlappningar i partitionerna visas fönstret med varningsmeddelande (figur 3). Läs och klicka på OK för att fortsätta.
Figur 3: Fönster med varningsmeddelande (visas endast om det finns kodningsöverlappningar i partitionerna).
När de väl är inlästa visas fem teckenpartitioner i huvudpanelen (figur 4). Anpassningen är uppdelad i en proteinkodande del och en icke-kodande del,och den kodande delen är uppdelad i kodonpositionerna 1, 2 och 3. Du måste ta bort partitionen ”coding” innan du fortsätter till nästa steg eftersom den hänvisar till samma nukleotider som partitionerna ”1stpos”, ”2ndpos” och ”3rdpos”. För att ta bort partitionen ”kodning” markerar du raden och klickar på knappen ”-” längst ner i tabellen. Du kan visa anpassningen genom att dubbelklicka på partitionen.
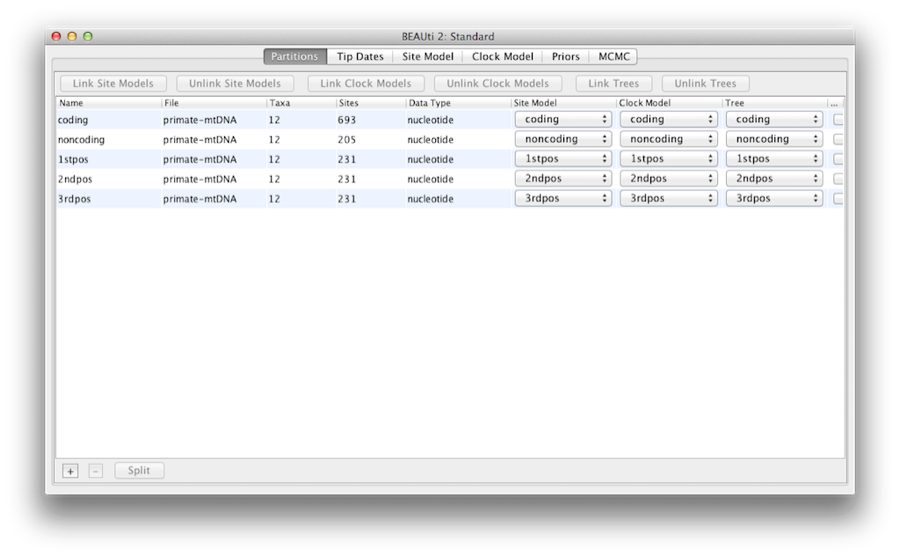
Figur 4: Skärmdump av fliken data i BEAUti. Denna och alla följande skärmdumpar togs på en Apple-dator med Mac OS X och kommer att se något annorlunda ut i andra operativsystem.
Länka/avlänka partitionsmodeller
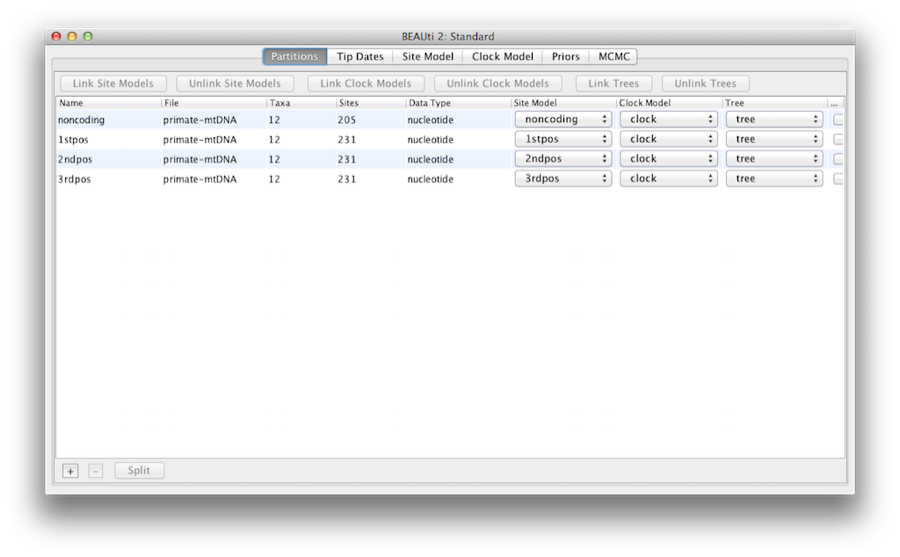
Figur 5: Skärmdump av fliken Partitioner i BEAUti efter det att klockmodellen och trädet länkats och döpts om.
Då sekvenserna är länkade (dvs. de kommer alla från det mitokondriella genomet som inte tros genomgå rekombination hos fåglar och däggdjur) har de samma härstamning, så partitionerna bör dela samma tidsträd i modellen. För enkelhetens skull kommer vi också att anta att partitionerna har samma utvecklingshastighet för varje gren, och därmed samma ”klockmodell”.Vi kommer att begränsa vår modellering av hastighetsheterogenitet till heterogenitet mellan olika platser inom varje partition, och vi kommer också att tillåta att partitionerna har olika genomsnittlig utvecklingshastighet.
Så här måste vi koppla ihop klockmodellen och trädet. I panelen Partitions väljer du alla fyra partitioner i tabellen (eller ingen, som standard påverkas alla partitioner) och klickar på knappen Link Trees och sedan på knappen Link Clock Models (se figur 5). Klicka sedan på den första rullgardinsmenyn i kolumnen Clock Model och byt namn på den delade klockmodellen till ”clock”. På samma sätt byter du namn på det delade trädet till ”tree”. Detta gör följande alternativ och genererade loggfiler lättare att läsa.
2.2 Inställning av substitutionsmodellen
Nästa steg är att ställa in substitutionsmodellen. Välj sedan fliken Site Models högst upp i huvudfönstret (vi hoppar över fliken Tip Dates eftersom alla taxa kommer från samtida prov). Detta kommer att avslöja inställningarna för den evolutionära modellen för BEAST. De tillgängliga alternativen beror på om uppgifterna är nukleotider eller aminosyror, binära uppgifter eller allmänna uppgifter. De inställningar som kommer att visas efter att ha laddat in primatens nukleotidanpassning kommer att vara standardvärdena för nukleotiddata, så vi måste göra några ändringar.
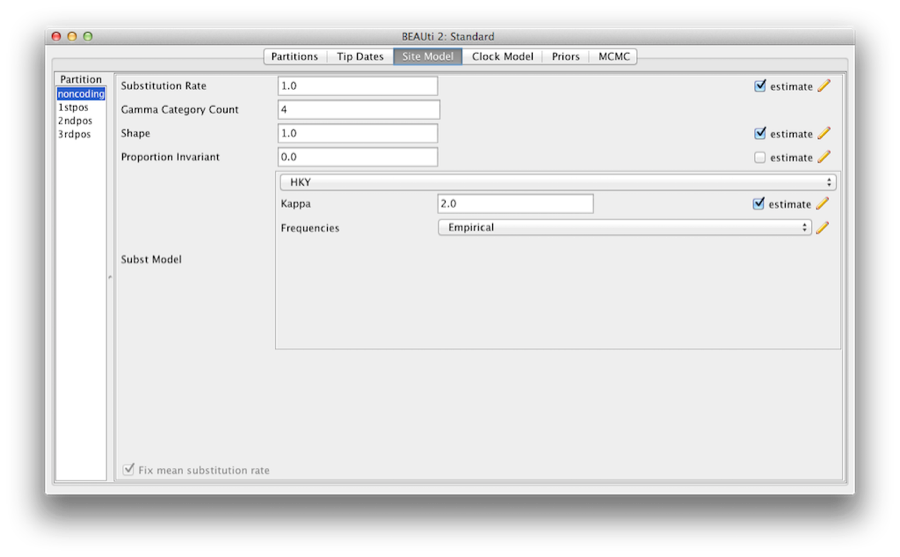
Figur 6: En skärmdump av fliken Site Model i BEAUti.
De flesta modellerna bör vara bekanta för dig. Ställ först in Gamma Category Count till 4 och kryssa sedan i rutan ”estimate” för Shape-parametern. Detta gör det möjligt att modellera hastighetsvariationer mellan platser i varje partition. Observera att 4 till 6 kategorier fungerar tillräckligt bra för de flesta dataset, medan det tar längre tid att beräkna fler kategorier utan någon större nytta. Vi lämnar posten Proportion invariant inställd på noll.
Välj sedan HKY från rullgardinsmenyn Subst Model. I idealfallet bör man välja en substitutionsmodell som passar data bäst för varje partition, men här använder vi för enkelhetens skull HKY för alla partitioner. Välj vidare Empirisk från rullgardinsmenyn Frekvenser. Detta kommer att fastställa frekvenserna till de proportioner som observerats i data (för varje partition individuellt, när vi väl har kopplat bort platsmodellerna). Detta tillvägagångssätt innebär att vi kan få en bra anpassning till data utan att explicit skatta dessa parametrar. Vi gör det här helt enkelt för att göra loggfilerna lite kortare och mer läsbara i senare delar av övningen.
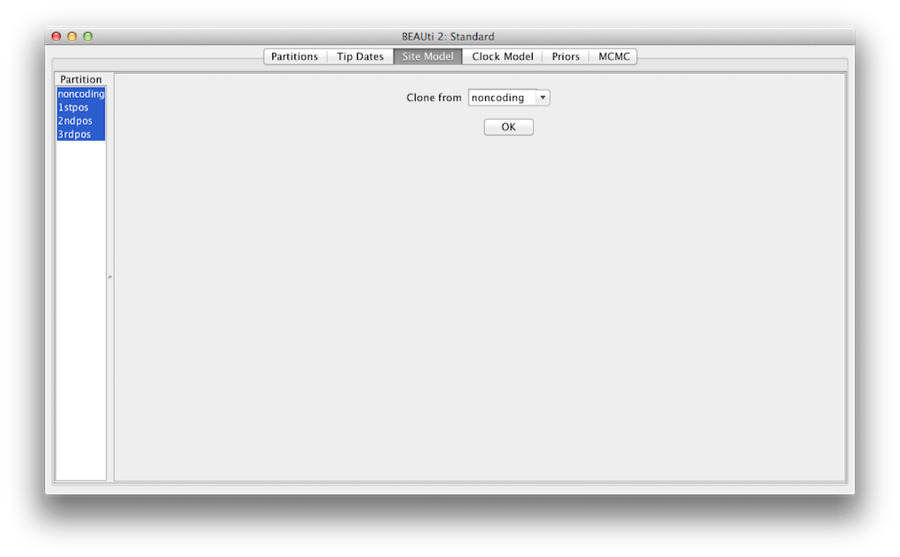
Figur 7: Klonkonfiguration från en platsmodell till andra.
Kryssa slutligen för rutan ”estimate” (uppskatta) för parametern Substitution rate (substitutionshastighet) och välj rutan Fix mean mutation rate (fixa medelmutationshastighet). Detta gör det möjligt för de enskilda partitionerna att få sina relativa hastigheter uppskattade för obundna platsmodeller (figur 6).
Håll slutligen in ”shift”-tangenten för att välja alla platsmodeller på vänster sida och klicka på OK för att klona inställningen från icke-kodning till 1stpos, 2ndpos och 3rdpos (figur 7). Gå igenom varje platsmodell, som du kan se är deras konfigurationer samma nu.
2.3 Inställning av klockmodellen
Nästa steg är att välja fliken Clock Models högst upp i huvudfönstret. Det är här vi väljer den molekylära klockmodellen. För den här övningen kommer vi att låta valet stå kvar på standardvärdet för en strikt molekylär klocka, eftersom dessa data är mycket klockliknande och inte behöver hastighetsvariation mellan grenar för att inkluderas i modellen.
För att testa om klocklikheten kan du (i) köra analysen med en avslappnad klockmodell och kontrollera hur mycket variation mellan hastigheterna som impliceras av data (se variationskoefficient för mer information om detta), eller(ii) utföra en modelljämförelse mellan en strikt och en avslappnad klocka med hjälp av pathsampling, eller (iii) använda en slumpmässig lokal klockmodell som uttryckligen tar hänsyn till om varje gren i trädet behöver sin egen grenhastighet.
2.4 Priorer
Fliken Priorer gör det möjligt att ange priorer för varje parameter i modellen. De modellval som görs i flikarna Site Model och Clock Model resulterar i att olika parametrar inkluderas i modellen, och dessa visas i fliken Priors (se figur 8).
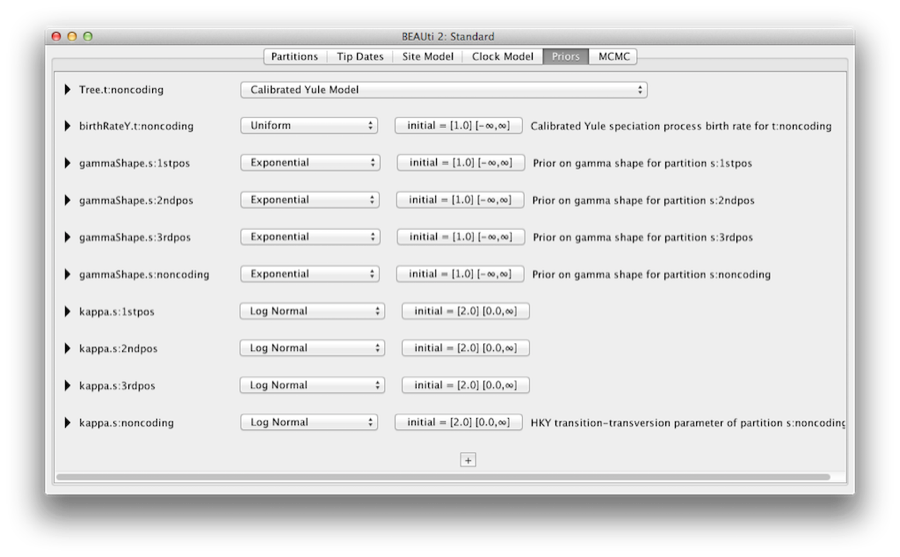
Figur 8: Skärmdump av fliken Priors i BEAUti.
Här anger vi också att vi vill använda den kalibrerade Yule-modellen som trädprioritet. Yule-modellen är en enkel modell för artspridning som i allmänhet är lämpligare när man betraktar sekvenser från olika arter. Välj Calibrated Yule Model från rullgardinsmenyn Tree prior.
2.4.1 Definiera kalibreringsnoden
Vi måste nu specificera en prioritetsfördelning på den kalibrerade noden, baserad på vårfossilkunskap. Detta kallas för att kalibrera vårt träd. Om du vill definiera en extra prioritet trycker du på den lilla +-knappen under listan över prioriteter. Om den inte är synlig i din vy kan du bläddra ner i panelen till botten för att hitta +-knappen. Du kommer att se en dialogruta där du kan definiera en delmängd av taxa i det fylogenetiska trädet. När du har skapat en uppsättning taxa kan du senare lägga till kalibreringsinformation för dess senaste gemensamma föregångare (MRCA).
Nämna taxauppsättningen genom att fylla i posten taxauppsättningsetikett. Kalla den human-chimp, eftersom den kommer att innehålla taxa för Homo sapiens och Pan. I listan nedan visas de tillgängliga taxa. Välj var och en av de två taxa i tur och ordning och tryck på pilknappen > >. (Figur 9).Klicka på OK och den nydefinierade taxauppsättningen kommer att läggas till i prioritetslistan.Eftersom detta är en kalibrerad nod som ska användas tillsammans med Calibrated Yule prior måste monofylikt upprätthållas, så markera kryssrutan Monophyletic. Detta kommer att begränsa trädets topologi så att grupperingen människa-chimpans hålls monofyletisk under MCMC-analysen.
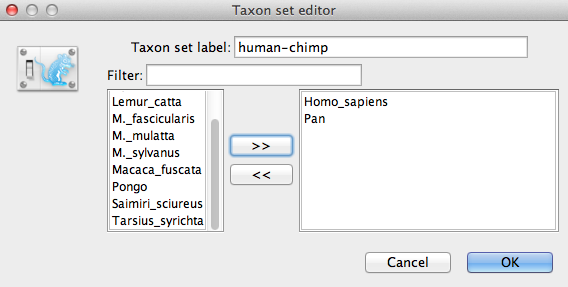
Figur 9: Redigerare för taxonuppsättningar i BEAUti.
För att koda kalibreringsinformationen måste vi ange en fördelning för MRCA för human-chimpans.Välj log-normalfördelningen från rullgardinsmenyn till höger om den nyligen tillagda human-chimp.prior. Klicka på den svarta triangeln så visas en graf över sannolikhetstäthetsfunktionen tillsammans med parametrar för log-normalfördelningen. vi kommer att ställa in M=1,78 och S=0,085 vilket kommer att specificera en fördelning som är centrerad vid cirka 6 miljoner år med en standardavvikelse på cirka 0,5 miljoner år. Detta ger ett centralt 95-procentigt sannolikhetsintervall som täcker 5-7 Mya. Detta motsvarar ungefär den nuvarande konsensusuppskattningen av datumet för den senaste gemensamma förfadern för människor och schimpanser (figur 10).
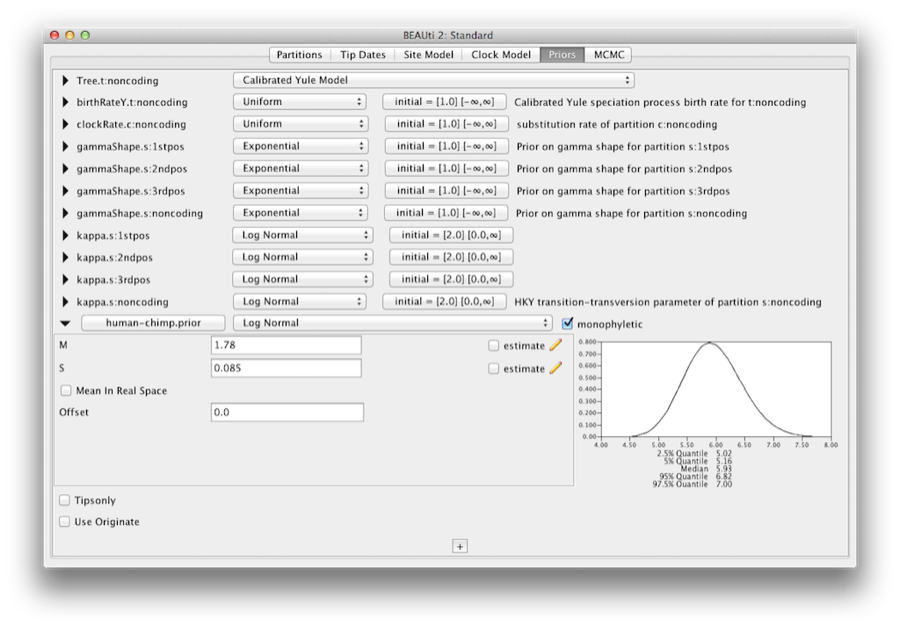
Figur 10: Skärmdump av alternativen för kalibreringsprioritet i panelen Priors i BEAUti.
Vi bör övertyga oss själva om att de priorer som visas i panelen priors verkligen återspeglar den förhandsinformation vi har om modellens parametrar. Slutligen kommer vi också att ange några diffusa ”icke-informativa” men korrekta priorer på den övergripande molekylära klockfrekvensen (clockRate) och artbildningsfrekvensen (birthRateY) i Yule-trädsprioriteten. För var och en av dem väljer du Gamma från rullgardinsmenyn och med hjälp av pilknappen expanderar du vyn för att visa parametrarna för Gamma-prioriteringen. För både klockfrekvensen och födelsefrekvensen för Yule ställ in parametern Alpha(shape) till 0,001 och parametern Beta(scale) till 1000 (figur 11).
Som standard har var och en av parametrarna för Gammas form en exponentiell prioritetsfördelning med medelvärde 1. Detta innebär (se figur 3.7) att vi förväntar oss en viss variation. Som standard har kappaparametrarna för HKY-modellen en lognormal(1,1,25) prioritetsfördelning, vilket i stort sett stämmer överens med empiriska belägg för intervallet av realistiska värden för övergångs-/omvandlingsbias. Dessa standardprioriteter behålls eftersom de är lämpliga för just denna analys.
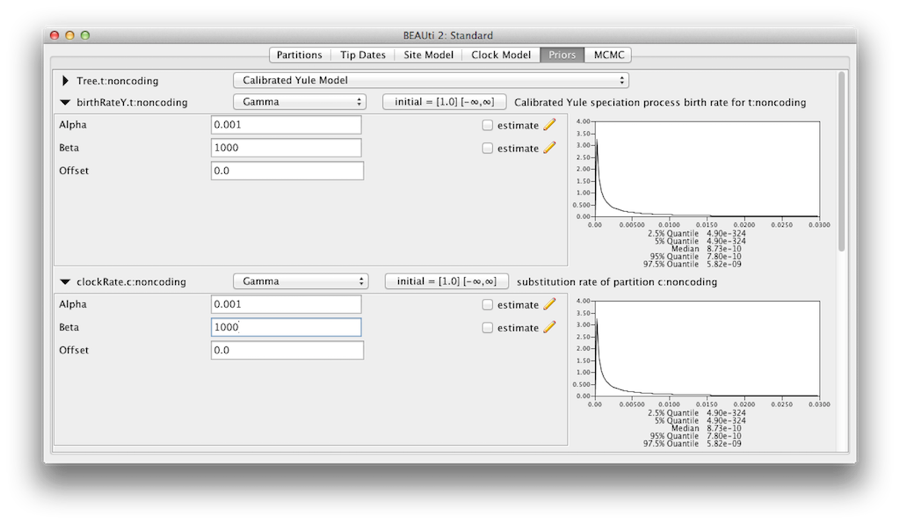
Figur 11: Gammaprioritet.
2.5 Inställning av MCMC-alternativ
Nästa flik, MCMC, ger mer generella inställningar för att styra längden på MCMC-körningen och filnamnen.
För det första har vi kedjelängden. Detta är antalet steg som MCMC kommer att göra i kedjan innan den avslutas. Hur lång denna kedja bör vara beror på datamängdens storlek, modellens komplexitet och kvaliteten på det svar som krävs. Standardvärdet 10 000 000 000 är helt godtyckligt och bör anpassas till storleken på din datamängd. För den här datamängden sätter vi kedjelängden till 6 000 000 000 eftersom detta kommer att köras ganska snabbt på de flesta moderna datorer (några minuter).
Fältet Store Every (lagra varje) bestämmer hur ofta tillståndet lagras i en fil. Att lagra tillståndet regelbundet är användbart i situationer där datormiljön inte är särskilt tillförlitlig och en BEAST-körning kan avbrytas. Om du har en lagrad kopia av det senaste tillståndet kan du återuppta kedjan i stället för att börja om från början, så att du inte behöver gå igenom inbränningen igen.Pre Burnin-fältet anger antalet prover som inte loggas i början av analysen. Vi låter fälten Store Every och Pre Burnin vara inställda på sina standardvärden. Nedan följer detaljerna för loggfilerna. Var och en kan utökas genom att klicka på den svarta triangeln.
Nästa alternativ anger hur ofta parametervärdena i Markovkedjan ska visas på skärmen och registreras i loggfilen.Skärmutgången är helt enkelt till för att övervaka programmets framsteg och kan ställas in på vilket värde som helst (om den ställs in för lågt kommer den stora mängden information som visas på skärmen att sakta ner programmet). För loggfilen bör värdet ställas in i förhållande till kedjans totala längd. Om proverna tas för ofta kommer det att resultera i mycket stora filer med liten extra nytta när det gäller analysens noggrannhet. Om provtagning sker för sällan kommer loggfilen inte att registrera tillräcklig information om parametrarnas fördelning. Du vill förmodligen sträva efter att inte lagra mer än 10 000 prover, så detta bör vara minst kedjelängd / 10 000.
För den här övningen kommer vi att ställa in frekvensen för spårlogg och trädlogg till 1 000 och för skärmlogg till 10 000. Vi kommer också att ange Primates.log som filnamn för spårloggfilen och Primates.trees som filnamn för trädloggfilen.Se till att filnamnet för skärmloggen lämnas tomt, annars skrivs inte skärmloggen till skärmen.
- Om du använder operativsystemet Windows föreslår vi att du lägger till suffixet .txt till båda dessa (alltså Primates.log.txt och Primates.trees.txt) så att Windows känner igen dem som textfiler.
2.6 Generering av BEAST XML-filen
Vi är nu redo att skapa BEAST XML-filen. För att göra detta väljer du alternativet Save (Spara) i menyn File (Fil). Kontrollera standardpriorerna och spara filen med ett lämpligt namn (vi brukar avsluta filnamnet med .xml, dvs. Primates.xml) Vi är nu redo att köra filen genom BEAST.
3 Kör BEAST
Figur 12: En skärmdump av BEAST.
Kör nu BEAST och när den frågar efter en inmatningsfil, ge din nyskapade XML-fil som indata. BEAST kommer sedan att köras tills den är klar med att rapportera information till skärmen. De faktiska resultatfilerna sparas på disken på samma plats som din inmatningsfil. Utmatningen på skärmen kommer att se ut ungefär så här:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Bemärk att det finns en del användbar information i början om anpassningarna och vilka trädsannolikheter som används. Dessutom nämns alla citat som är relevanta för analysen i början av körningen, vilket lätt kan kopieras tillomanuskript som rapporterar om analysen. Därefter följer rapportering av kedjan,vilket ger en viss återkoppling i realtid om kedjans framskridande.
I slutet skrivs en operatörsanalys ut, som listar alla operatörer som använts i analysen tillsammans med hur ofta operatören prövades, accepterades och förkastades (se kolumnerna #total, #accept och #reject respektive). Acceptansgraden är den andel av gångerna en operatör accepteras när den väljs ut för att göra ett förslag. I allmänhet tyder en hög acceptansgrad, t.ex. över 0,5, på att förslagen är konservativa och att de inte utforskar parameterutrymmet på ett effektivt sätt. Å andra sidan tyder en låg acceptansgrad på att förslagen är för aggressiva och nästan alltid resulterar i ett tillstånd som förkastas på grund av dess låga posterior.Både för höga och för låga acceptansgrader resulterar i låga ESS-värden. En acceptansfrekvens på 0,234 är målet (baserat på mycket begränsade bevis som tillhandahålls av ) för många (men inte alla) operatörer som implementerats i BEAST.
Vissa operatörer har en inställningsparameter, till exempel skalfaktorn för ascale-parametern. Om den slutliga acceptansgraden inte ligger nära målet föreslår BEAST ett nytt värde för inställningsparametern, vilket skrivs ut i operatörsanalysen. I det här fallet är alla acceptansfrekvenser bra för de operatörer som har inställningsparametrar. Operatörer utan inställningsparametrar inkluderar wideexchange- och Wilson-Balding-operatörerna för denna analys. Båda dessa operatörer försöker ändra trädets topologi med stora steg, men eftersom uppgifterna till övervägande del stöder en enda topologi förkastas dessa radikala förslag nästan alltid.
4 Analysera resultaten
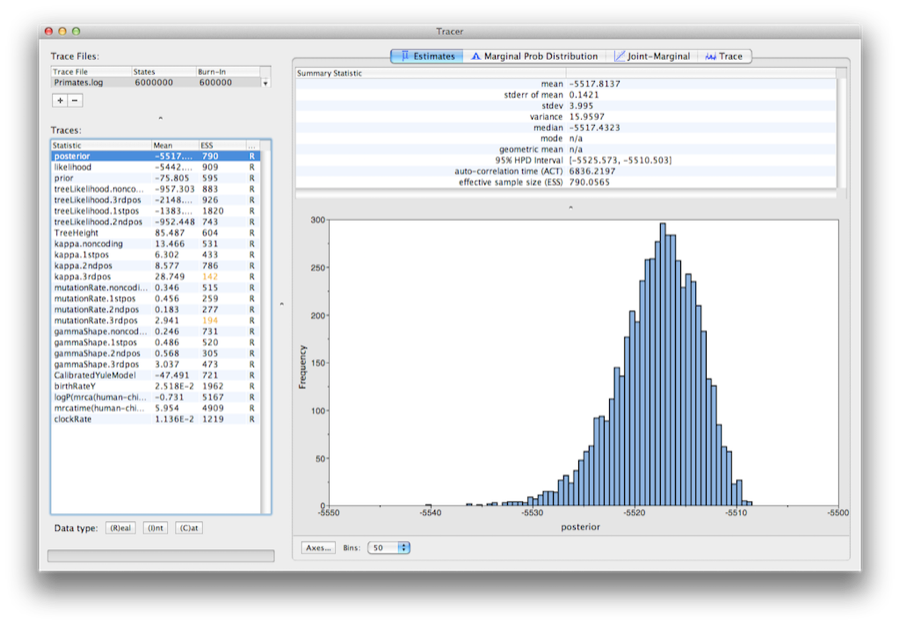
Figur 13: En skärmdump av Tracer v1.6.
Kör programmet som heter Tracer för att analysera resultatet av BEAST. När huvudfönstret har öppnats väljer du Import Trace File… från menyn File och väljer den fil somBEAST har skapat och som heter Primates.log (Figur 13).
Håll i minnet att MCMC är en stokastisk algoritm, så de faktiska siffrorna kommer inte att vara exakt desamma som de som avbildas i figuren.
På vänster sida finns en lista över de olika kvantiteter som BEAST har loggat till filen. Det finns spår för posterior (detta är den naturliga logaritmen av produkten av trädets sannolikhet och prioritetsdensiteten) och de kontinuerliga parametrarna. Genom att välja en spårning till vänster visas analyser för denna spårning till höger, beroende på vilken flik som valts. Vid första öppnandet väljs ”posterior”-spåret och olika statistiska uppgifter för detta spår visas under fliken Estimates (uppskattningar).I den övre högra delen av fönstret finns en tabell med beräknad statistik för det valda spåret.
Välj parametern clockRate i den vänstra listan för att titta på den genomsnittliga utvecklingshastigheten (medelvärde över hela trädet och alla platser). Tracer kommer att rita ett histogram (marginal posterior) för den valda statistiken och även ge dig sammanfattande statistik som t.ex. medelvärde och median. 95 % HPD står för highest posterior density-intervallet och representerar det mest kompakta intervallet för den valda parametern som innehåller 95 % av den efterföljande sannolikheten. Det kan fritt ses som en Bayesiansk analog till ett konfidensintervall. Parametern TreeHeight ger den marginella posteriora fördelningen av åldern på roten till hela trädet.
Välj parametern TreeHeight och Ctrl-klicka sedan på mrcatime(human-chimp) (Command-click på Mac OS X). Detta kommer att visa en visning av åldern på roten och den kalibrerings-MRCA som vi angav tidigare i BEAUti. Du kan kontrollera att divergensen som vi använde för att kalibrera trädet(mrcatime(human-chimp)) har en efterföljande fördelning som stämmer överens med den prioritetsfördelning som vi specificerade (figur 14).
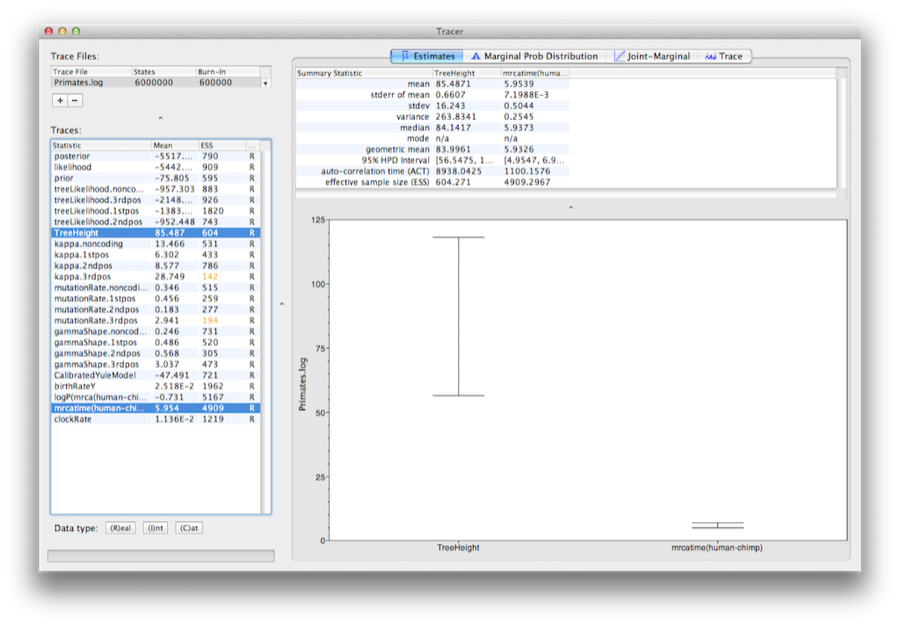
Figur 14: Skärmdump av de 95 % HPD-intervallerna för rothöjden och den användardefinierade (human-chimp) MRCA i Tracer.
5 Marginella posteriora skattningar
För att visa de relativa hastigheterna för de fyra partitionerna väljer du parametern mutationRate för var och en av de fyra partitionerna och väljer fliken Marginaltäthet i Tracer. 15 visar marginaltätheterna för de relativa substitutionshastigheterna. Plotten visar att kodonpositionerna 1 och 2 har väsentligt olika hastigheter (0,456 mot 0,183) och båda är mycket långsammare än kodonposition 3 med en relativ hastighet på 2,941. Den icke-kodande partitionen har en hastighet som ligger mellan kodonpositionerna 1 och 2 (0,346). Sammantaget tyder detta resultat på ett starkt renande urval i både de kodande och icke-kodande regionerna i anpassningen.
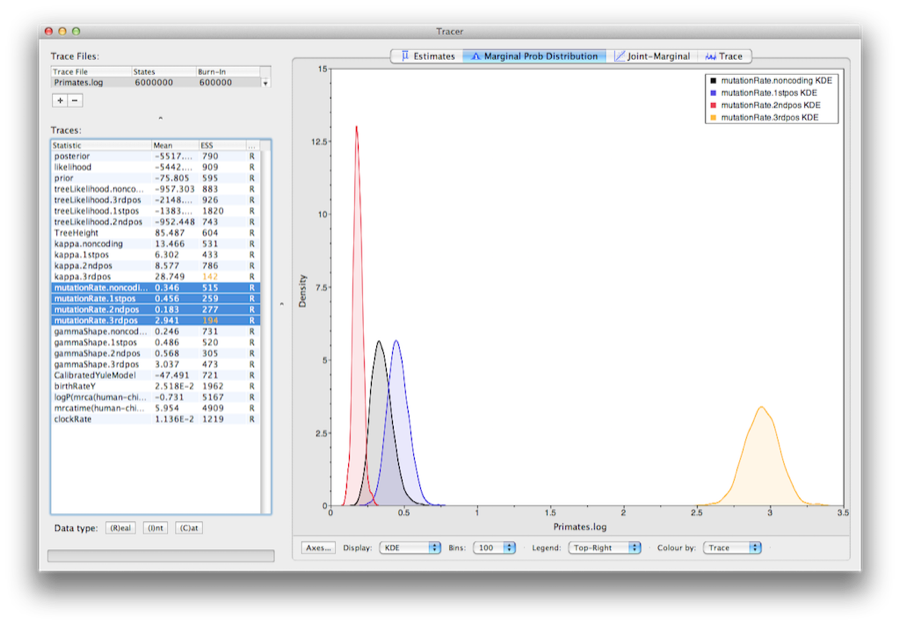
Figur 15: En skärmdump av de marginella posteriora tätheterna av de relativa substitutionshastigheterna för de fyra partitionerna (i förhållande till den platsviktade medelhastigheten).
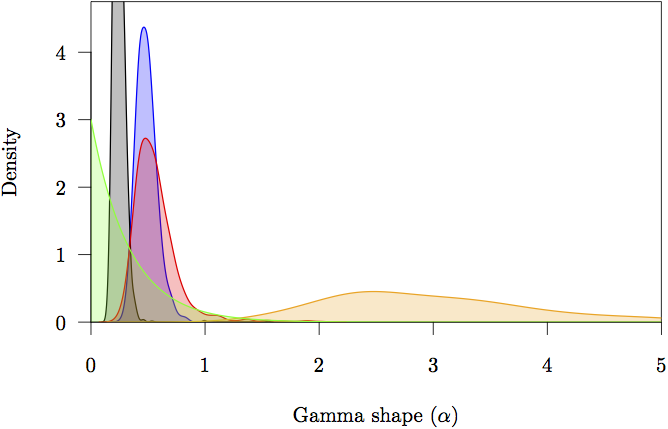
Figur 16: De marginella prior- och posteriortätheterna för formparametrarna (α). Prioriteten är grå. Posteriortäthetsskattningen för varje partition visas också: icke-kodande (orange) och första (röd), andra (grön) och tredje (blå) kodonpositioner.
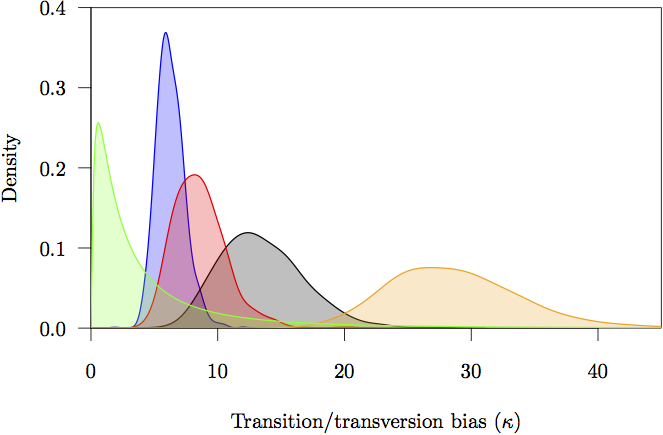
Figur 17: Marginalprioriteten och posteriortätheterna för parametrarna för övergångs-/övergångsbias (κ). Prioriteten är grå. Posteriortäthetsskattningen för varje partition visas också: icke-kodande (orange) och första (röd), andra (grön) och tredje (blå) kodonpositioner.
Frågor
Vilken är den uppskattade hastigheten för den molekylära evolutionen för detta genträd (inkludera HPD-intervallet på 95 %)?
Vilka felkällor ingår i denna uppskattning?
Hur gammal är trädets rot (ange medelvärdet och HPD-intervallet på 95 %)?
6 Upprättande av en uppskattning av det fylogenetiska trädet
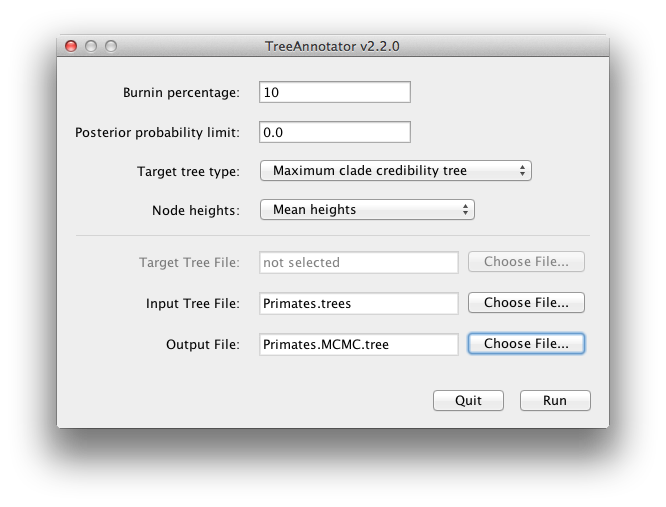
BEAST framställer också ett posteriort urval av fylogenetiska tidsträd tillsammans med sitt urval av parameterskattningar. Dessa måste sammanfattas med hjälp av programmet TreeAnnotator. Detta kommer att ta upp uppsättningen träd och hitta det bäst stödda trädet. Det kommer sedan att kommentera detta representativa sammanfattande träd med medelåldrarna för alla thenodes och motsvarande 95 % HPD-områden. Det kommer också att beräkna den efterföljande kladsannolikheten för varje nod. Kör programmet TreeAnnotator och ställ in det enligt figur 18.
Figur 18: Skärmdump av TreeAnnotator.
Burnin är antalet träd som ska tas bort från början av urvalet. Till skillnad från Tracer, som anger antalet steg som burnin, måste du i TreeAnnotator ange det faktiska antalet träd. För den här körningen angav du en kedjelängd på 6 000 000 000 steg med provtagning vart 1 000:e steg. Trädfilen kommer alltså att innehålla 6 000 träd och därför måste du ange en 10 % brännpunkt i det översta textfältet.
Objektet Posterior probability limit anger en gräns som innebär att om en nod hittas med mindre än denna frekvens i urvalet av träd (dvs. har en posterior sannolikhet som är mindre än denna gräns), kommer den inte att kommenteras. Standardvärdet 0,5 innebär att endast noder som förekommer i majoriteten av träden kommer att kommenteras. Ställ in detta till noll för att kommentera alla noder.
Målträdstypen anger den trädtopologi som kommer att kommenteras. Du kan antingen välja ett specifikt träd från en fil eller be TreeAnnotator att hitta ett träd i ditt prov. Standardalternativet, Maximum clade credibility tree, hittar trädet med den högsta produkten av den efterföljande sannolikheten för alla dess noder.
För nodhöjder är standardalternativet Common Ancestor Heights, som beräknar höjden på en nod som medelvärdet av MRCA-tiden för alla par av noder i kladen. För träd med stor osäkerhet i topologin och därmed många klader med lågt stöd kan vissa andra metoder resultera i träd med negativa grenlängder. I den här analysen är stödet för alla klader i det sammanfattande trädet myckethögt, så detta är inget problem här.Välj medelhöjder för nodhöjder. Detta ställer in höjderna (åldrarna) för varje nod i trädet till medelhöjden i hela urvalet av träd för den kladen.
För inmatningsfilen väljer du trädfilen som BEAST skapade och väljer en fil för resultatet (här kallade vi den Primates.MCC.tree). Tryck nu på Kör och vänta tills programmet är klart.
7 Visualisering av trädskattningen
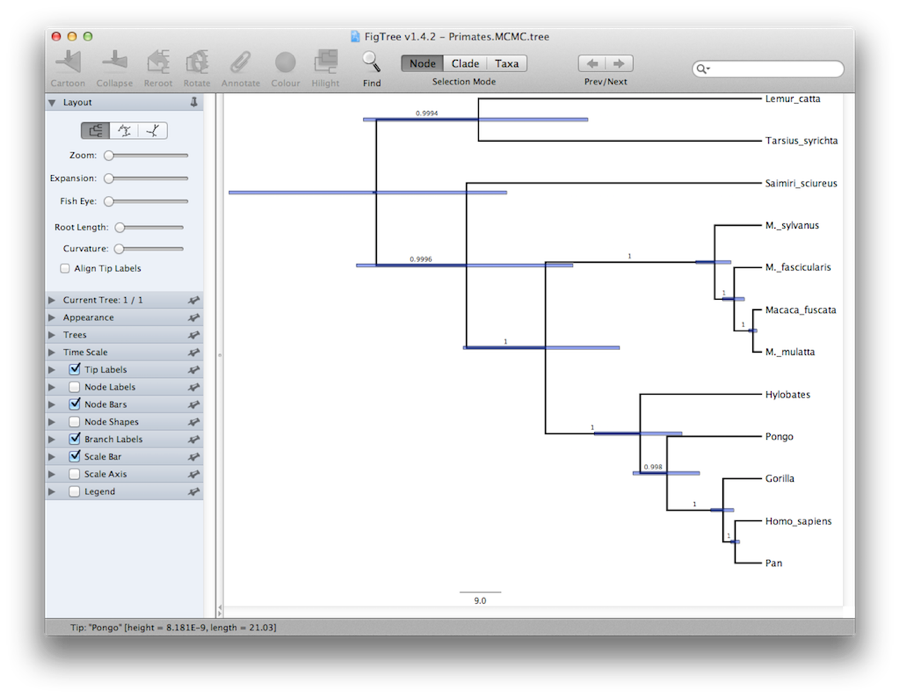
Slutligt kan vi visualisera trädet i ett annat program som heter FigTree. Kör det här programmet och öppna filen Primates.MCC.tree med hjälp av kommandot Open i menyn File. Trädet bör visas.Du kan nu prova att välja några av alternativen i kontrollpanelen till vänster. Först av allt, förbrukar du Trees-alternativet i panelen och kryssar i Order nodes och väljer Ordering by decreasing (ordna genom att minska). Försök att välja Node Bars för att få fram felmarginaler för nodens ålder. Slå också på Branch Labels och välj posterior för att getit att visa den posteriora sannolikheten för varje nod. Om du använder en icke strikt klockmodell kan du under Appearance också säga åt FigTree att färga grenarna efter hastigheten. du bör få något som liknar figur 19.
Figur 19: Skärmdump av FigTree och DensiTree.
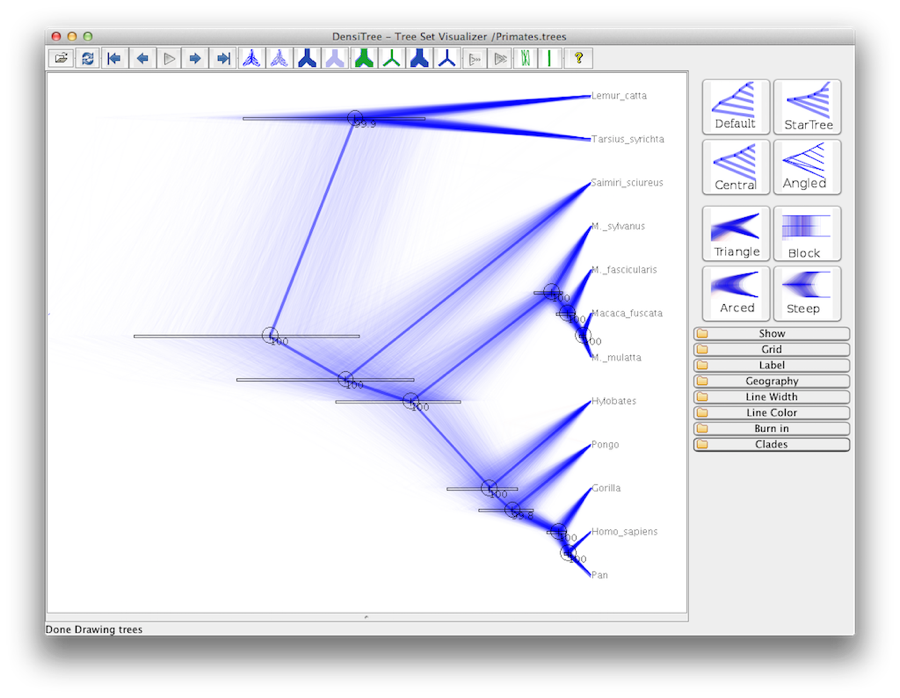
En alternativ vy av trädet kan göras med DensiTree, som är en del av Beast 2. Fördelen med DensiTree är att det kan visualisera både osäkerheten i nodhöjder och osäkerheten i topologin. för denna speciella datauppsättning finns den dominerande topologin i mer än 99 % av proven. Så vi drar slutsatsen att den här analysen resulterar i en mycket hög konsensus om topologin (figur 19).
Frågor
- Dennasi, skiljer sig evolutionshastigheten väsentligt mellan de olika linjerna i trädet?
- DensiTree har en kladebalk (menyfönster/View clade toolbar) för att visa information om klader.
Vad är stödet för kladen?
- Du kan bläddra bland topologierna i DensiTree med hjälp av menyn Browse.Den mest populära topologin har ett stöd på över 99 %.
Vad är stödet för den näst mest populära topologin?
- Under hjälpmenyn visar DensiTree viss information.
Hur många topologier finns i träduppsättningen?
8 Jämförelse av dina resultat med prioriteten
Det är en bra idé att köra om analysen medan du samplar från prioriteten för att försäkra dig om att interaktioner mellan priorer inte påverkar din prioritetsinformation. Interaktionen mellan priors kan vara problematisk, särskilt när man använder kalibreringar eftersom det innebär att man lägger flera priors på trädet.
Med hjälp av BEAUti ställer du in samma analys, men under MCMC-alternativen väljer du alternativet Sample from prior only (endast provtagning från prior). På så sätt kan du visualisera den fullständiga prioritetsfördelningen i avsaknad av dina sekvensdata. Sammanfatta träden från den fullständiga prioritetsfördelningen och jämför sammanfattningen med det efterföljande sammanfattningsträdet.
Divergenstidsuppskattning med hjälp av ”noddatering” av den typ som beskrivs i det här kapitlet har tillämpats för att besvara en mängd olika frågor inom ekologi och evolution. Till exempel har node dating med fossil använts för att bestämma artdiversiteten hos cycader, analysera utvecklingshastigheten hos blommande växter och undersöka ursprunget till cyanobakterier i varma och kalla öknar.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, m.fl, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond och Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), nr 1, 114. A Gelman, G Roberts och W Gilks, Efficient metropolis jumping hules, Bayesian statistics 5 (1996), 599-608. Joseph Heled och Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little och S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian och Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith och Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008), no. 5898, 86-89.
Detta dokument har översatts från LATEX avHEVEA.