John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Le prove crescenti evidenziano l’intima relazione tra il diabete di tipo II (T2D) e la malattia di Alzheimer (AD). È importante notare che queste due malattie condividono una serie di somiglianze patologiche, tra cui l’accumulo di amiloide, lo stress ossidativo, l’infiammazione e la morte cellulare. Ad oggi, le terapie farmacologiche per AD e T2D sono carenti e c’è un bisogno cruciale di scoprire e sviluppare nuove terapie per queste malattie. Una serie di studi sull’uomo e sui roditori ha dimostrato che l’integrazione dell’ormone metabolico è molto utile per migliorare la funzione cognitiva e la salute metabolica generale sia nel T2D che nel MA. L’ormone pancreatico amilina è emerso come una componente cruciale dell’eziologia della malattia sia di T2D che di AD, anche se il ruolo esatto che l’amilina gioca in queste malattie non è ancora ben compreso. Qui, rivediamo criticamente la letteratura attuale che utilizza l’amilina umana o il suo analogo sintetico, la pramlintide, così come gli antagonisti del recettore dell’amilina per il trattamento dell’AD.
Introduzione
La malattia di Alzheimer (AD) è una malattia neurodegenerativa progressiva e debilitante caratterizzata dall’accumulo di placche di amiloide-beta (Aβ) e grovigli neurofibrillari composti da tau1 iperfosforilato. L’accumulo di questi peptidi patologici contribuisce a deficit nelle funzioni esecutive come l’apprendimento e la memoria, l’umore, l’affetto, ecc. e presenta un carico sostanziale per il paziente e i caregiver. L’incidenza del MA sta aumentando ad un ritmo allarmante negli Stati Uniti, con una stima di 5,5 milioni di americani che vivono con il MA a partire dal 2017 e questo numero dovrebbe triplicare entro il 20502. Inoltre, il costo dell’assistenza e del trattamento dei pazienti con AD supera attualmente i 200 miliardi di dollari all’anno e si prevede solo un aumento3. Mentre AD è chiaramente un problema monumentale all’interno degli Stati Uniti e oltre, le opzioni di trattamento rimangono molto limitate4. Sono state condotte molte sperimentazioni farmacologiche con una vasta gamma di approcci mirati, ma attualmente ci sono solo sei farmaci approvati dalla FDA per il MA e sono solo trattamenti sintomatici5, 6. Ad oggi, la maggior parte degli agenti farmacologici sviluppati hanno mirato specificamente alla patologia Aβ o tau, ma nessuno ha avuto successo nel cancellare o prevenire la patologia4. Come tale, c’è un bisogno fondamentale di sviluppare terapie valide e trattamenti preventivi per AD.
Age-related (sporadico) AD è una complicata malattia multifattoriale, avendo numerose influenze genetiche e ambientali. L’ambiente e lo stile di vita sono fortemente implicati nello sviluppo del MA sporadico; fattori come la dieta7-9, l’obesità8-10, la sindrome metabolica7, il diabete di tipo II (T2D)9, 11 e le malattie cardiovascolari12 sono stati tutti implicati nella causalità del MA. Di fondamentale importanza, i tassi di obesità e diabete sono in rapido aumento in parallelo con AD12, 13. Anche se il rapporto tra obesità e AD è poco chiaro, ci sono prove che l’obesità di mezza età gioca un ruolo nello sviluppo di AD10. Ancora più importante, l’obesità è comunemente accompagnata da una serie di altre malattie, tra cui malattie cardiovascolari, ipertensione, dislipidemia, T2D, ictus, ecc.14. L’incidenza di T2D è in rapido aumento, con il CDC stima che circa 30,3 milioni di persone (1 su 10 adulti) negli Stati Uniti hanno il diabete e un impressionante 84,1 milioni (1 su 3 adulti) hanno prediabete, la maggior parte dei quali non sono consapevoli della loro condizione. Inoltre, a causa di una diminuzione su larga scala dell’attività fisica che è accompagnata da un simultaneo aumento dell’assunzione di cibo e dieta povera, i tassi di obesità, T2D, sindrome metabolica e malattie cardiovascolari sono solo proposti per aumentare a una stima di 600 milioni di casi di T2D in tutto il mondo entro 203515.
Il corpo di prove che implicano la funzione metabolica e la malattia nel processo di declino cognitivo e invecchiamento è sostanziale16, 17. Per esempio, circa il 70% delle persone con diagnosi di T2D riporta un deterioramento cognitivo e un numero sostanziale di pazienti T2D sviluppa in seguito la demenza16, 18-21. Gli individui con diagnosi di T2D per almeno cinque anni hanno un rischio significativamente aumentato di sviluppare AD rispetto a quelli che hanno sofferto di T2D per meno di cinque anni17. Insieme questi dati suggeriscono che la crescente prevalenza di T2D nella popolazione può contribuire all’aumento dei tassi di AD.
T2D è inizialmente caratterizzato da un alto livello di glucosio e insulina nel sangue, che porta a iperinsulinemia; importante, l’amilina, un piccolo ormone metabolico prodotto dalle cellule β-islet del pancreas, è co-packaged e co-secretato con insulina ed è quindi iperprodotto in T2D.22. È importante notare che ci sono una serie di caratteristiche patologiche che sono presenti sia nella T2D che nell’AD: 1) diminuzione del metabolismo cerebrale e resistenza agli ormoni metabolici 2) patologia amiloide 3) stress ossidativo (OS) e infiammazione. L’iperinsulinemia cronica e l’iperamilinemia portano a una serie di problemi fisiologici: l’iperinsulinemia cronica porta all’insulino-resistenza del sistema22, all’alterazione del trasporto dell’insulina attraverso la barriera emato-encefalica (BBB)23, 24 e quindi alla diminuzione della segnalazione dell’insulina nel cervello25. La perdita di segnalazione dell’insulina nel cervello è associata a una serie di caratteristiche patologiche legate all’AD, tra cui l’aumento della produzione di Aβ, la fosforilazione della tau e la neuroinfiammazione.
Inoltre, l’amilina condivide caratteristiche patologiche simili con l’Aβ ad alte concentrazioni26 e può essere un percorso comune tra le due malattie. Per esempio, le fibrille di amilina sono state trovate nel pancreas del 95% dei pazienti T2D27-29 e causano una serie di interruzioni fisiologiche tra cui l’afflusso aberrante di Ca2+, l’aumento della secrezione di citochine pro-infiammatorie30,31 e, infine, la perdita di cellule β-isletarie32. Inoltre, l’amilina attraversa facilmente la BBB e forma fibrille di amilina e placche miste con Aβ all’interno del cervello e può essere responsabile della patologia simile all’AD e dell’inseminazione di Aβ nella T2D33-35. L’amilina è nota per influenzare il potenziamento a lungo termine (LTP) nell’ippocampo e può avere un’influenza innata sulla funzione cognitiva nel cervello36-39. Tuttavia, se l’amilina è un insulto tossico in queste malattie o se la sua perdita funzionale attraverso l’aggregazione o la perdita tardiva delle cellule β nella T2D contribuisce allo sviluppo di un AD rimane poco chiaro.
La dicotomia di segnalazione dell’amilina
C’è ancora molto dibattito sul coinvolgimento del recettore dell’amilina (AMYR) e della segnalazione dell’amilina nella progressione della malattia e nell’eziologia della T2D e del AD. Il corpo della ricerca volto a discernere questa relazione si sta rapidamente espandendo. Tutte le ricerche pertinenti hanno costantemente dimostrato che la modulazione della segnalazione dell’amilina influisce sulla patologia legata all’AD. La natura di questa relazione, tuttavia, deve ancora essere concretamente chiarita. Diversi gruppi hanno prodotto dati convincenti che suggeriscono che la segnalazione di amilina è benefico nella prevenzione della patologia AD-correlata e deficit cognitivi sia in vivo che in vitro40-44. Soprattutto, pramlintide, una forma ricombinante non aggregante di amilina, utilizzato in combinazione con terapie insuliniche per il trattamento del diabete e migliora il controllo glicemico, riduce il peso corporeo, e riduce i marcatori sierici di OS45-47 mostra anche la promessa come un terapeutico AD. Ad oggi, tuttavia, non ci sono stati studi clinici che hanno mirato a utilizzare amilina o pramlintide come agente terapeutico nel trattamento della demenza. Prove evidenti da studi su roditori suggeriscono che il trattamento cronico con amilina umana o pramlintide pone un forte beneficio terapeutico nel ridurre la patologia legata all’AD; l’integrazione di amilina/pramlintide riduce i livelli di Aβ solubile, il carico della placca, la fosforilazione della tau, la neuroinfiammazione e l’OS, migliorando anche la cognizione40-42,44. I dati di cui sopra suggeriscono che una perdita di segnalazione innata dell’amilina nel SNC a causa dell’aggregazione dà luogo a un aumento del rischio per lo sviluppo di AD ed è trattata in modo più dettagliato in Grizzanti et al. 201848.
Al contrario, gli studi mostrano anche che l’amilina umana e Aβ hanno effetti tossici simili e che questi effetti tossici possono essere alleviati utilizzando l’antagonista AMYR36-39,49. Ad esempio, i dati mostrano che il trattamento in vivo con antagonisti AMYR produce benefici fisiologici molto simili al trattamento con amilina o pramlintide. Il trattamento di topi AD TgCRND8 con AC253, un antagonista AMYR, o la sua controparte ciclica cAC253 riduce la neuroinfiammazione, i livelli di Aβ solubile e il carico della placca, migliorando anche la cognizione50. Allo stesso modo, in vitro / ex-vivo studi dimostrano che l’amilina umana a basse dosi o Aβ provoca interruzioni in LTP e che questi deficit sono bloccati da AC253 o pramlintide38,39, e dosi più elevate di amilina umana / oligomeri di amilina sono associati con incontrollato afflusso di Ca2 +, che è fortemente legato alla morte cellulare26,32. Insieme, questi dati supportano una funzione tossica di oligomeri di amilina e quindi un potenziale meccanismo terapeutico per il blocco AMYR. Al contrario, altri hanno dimostrato che gli effetti benefici dell’amilina possono essere bloccati usando AC25341. Così, il potenziale terapeutico del trattamento o dell’inibizione dell’amilina rimane poco chiaro e mette in evidenza la natura complessa e dicotomica degli amiloidi nel cervello e nella periferia.
Piecing Together the Puzzle
Ci sono una serie di buchi nella letteratura attuale che devono essere riempiti per dare un quadro più completo della storia dell’amilina: 1) la natura del sistema innato dell’amilina e la segnalazione dell’amilina nel cervello 2) le capacità di segnalazione di Aβ e pramlintide attraverso i tre principali recettori AMYR e correlati 3) i meccanismi terapeutici attraverso i quali l’inibizione di amilina/pramlintide o AMYR è mediata. In primo luogo, nuovi dati interessanti dimostrano che l’AMYR non è solo coinvolto nella segnalazione, ma anche nel trasporto del ligando attraverso la BBB. L’AMYR è un recettore eterodimero che è composto da un recettore della calcitonina e da una proteina che modifica l’attività del recettore (1-3)51. A tal fine, un knockdown globale del 50% del recettore della calcitonina (un componente chiave del AMYR) ha ridotto significativamente la quantità di AC253 trovato nel cervello50, indicando che AMYR situati nella BBB sono coinvolti nel trasporto di questi ligandi nel cervello e può anche essere coinvolto nel shuttling amilina e pramlintide dentro / fuori del cervello. L’esistenza di questi meccanismi di trasporto nella BBB suggerisce che l’amilina ha probabilmente una funzione fisiologica innata nel cervello, poiché il suo trasporto nel cervello è strettamente controllato. Tuttavia, come la segnalazione dell’amilina o la sua mancanza porti alle caratteristiche patologiche dell’AD e se l’AMYR sia il veicolo attraverso il quale l’Aβ media i suoi effetti tossici rimane ancora poco chiaro.
In seguito, esistono prove contrastanti riguardo alla relazione tra Aβ e l’AMYR. Anche se diversi studi dimostrano chiaramente che l’amilina umana e l’Aβ hanno effetti simili sul LTP nel SNC e l’uso di inibitori AMYR migliora questi effetti deleteri36-39, altre prove suggeriscono che l’Aβ (1-42) è incapace di segnalare attraverso qualsiasi AMYR per evocare qualsiasi tipo di risposta cAMP in un’ampia varietà di concentrazioni52. È possibile che Aβ attivi diverse cascate di segnalazione attraverso l’interazione con l’AMYR o semplicemente agisca come un inibitore competitivo inerte, ma questo deve ancora essere dimostrato.
Inoltre, uno studio separato ha dimostrato che l’amilina oligomerica media i suoi effetti tossici direttamente attraverso l’AMYR e indirettamente attraverso TRPV4, un canale cationico non selettivo26. Basse concentrazioni di amilina umana evocare una risposta Ca2 + che è mediata attraverso il suo recettore nativo. Tuttavia, a concentrazioni più elevate, amilina umana forma oligomeri e attiva la segnalazione aberrante che si traduce in attivazione dei canali TRVP4 e permette l’afflusso incontrollato di cationi, in particolare Ca2 +. Il blocco farmacologico dell’AMYR e TRPV4 dimostra che entrambi i recettori sono necessari per l’amilina umana oligomerica per indurre i suoi effetti tossici Ca2+26. Come tale, è probabile che Aβ media i suoi effetti tossici sul AMYR in modo simile, anche se questi dati non esistono ancora. L’afflusso incontrollato di Ca2+ è legato a una serie di fenomeni patologici, tra cui il rilascio vescicolare incontrollato, la disfunzione OS e mitocondriale, l’apoptosi, ecc. A tal fine, è probabile che la disfunzione cellulare e lo sviluppo di ulteriore patologia simile all’AD che deriva dalla segnalazione tossica dell’amiloide sia mediata sia dall’AMYR che dal TRPV4. Come tale, è necessario discernere le cascate di segnalazione che modulano la relazione tra l’AMYR e TRVP4. Inoltre, gli esperimenti farmacologici sono garantiti che utilizzano Aβ e pramlintide su una vasta gamma di dosi per determinare gli effetti di Aβ e pramlintide sulle correnti di Ca2+, LTP, produzione di cAMP, e altre cascate di segnalazione per determinare le loro capacità di segnalazione. Questi esperimenti contribuiranno a riempire alcuni dei vuoti nella letteratura attuale per quanto riguarda l’AMYR e il suo coinvolgimento negli stati di malattia (Figura 1).
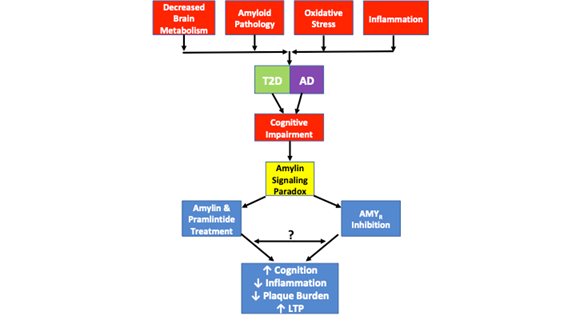
La Figura 1. raffigura il paradosso di segnalazione dell’amilina e le somiglianze patologiche osservate in T2D e AD. Diminuzione del metabolismo cerebrale, patologia amiloide, stress ossidativo e infiammazione sono tutte caratteristiche patologiche comuni osservate in entrambe le malattie. Mentre non tutti i casi di T2D o AD includono ognuna di queste caratteristiche patologiche, ogni caso presenta un deterioramento cognitivo. Il paradosso di segnalazione dell’amilina entra in gioco in quanto gli studi hanno dimostrato che sia l’inibizione dell’AMYR che l’agonismo dell’AMYR tramite l’amilina e il trattamento con pramlintide si traducono in un miglioramento della cognizione, una diminuzione dell’infiammazione, una diminuzione del carico della placca e un aumento dell’LTP. I meccanismi di segnalazione governati da agonismo AMYR e antagonismo AMYR devono ancora essere completamente chiariti. Mentre la segnalazione dell’amilina è tradizionalmente associata alla segnalazione di cAMP e PKA, non è chiaro se anche altre cascate sono attivate dall’amilina/pramlintide. Inoltre, non è chiaro se gli antagonisti AMYR, gli oligomeri di amilina o il segnale Aβ attraverso l’AMYR o se ci sono somiglianze o diafonie tra tutti questi ligandi AMYR. Come tale, una serie di esperimenti proposti in questa recensione aiuterà a chiarire ulteriormente la vera natura dell’AMYR.
Conclusioni
L’attuale disparità per quanto riguarda il ruolo della segnalazione dell’amilina nel cervello dimostra la necessità essenziale di chiarire ulteriormente il coinvolgimento dell’amilina sia nel MA che nel T2D. Nel T2D, è probabile che nelle prime fasi della malattia, l’amilina inonda il cervello, forma oligomeri, induce una segnalazione aberrante attraverso il suo recettore nativo, e recluta TRPV4 per indurre l’afflusso patologico di Ca2+ che si traduce in una diffusa disfunzione neuronale che si manifesta come OS, rilascio vescicolare incontrollato e disfunzione interneuronale, infiammazione e conseguente morte cellulare. Questo meccanismo può essere responsabile della transizione iniziale dal cervello sano all’invecchiamento del cervello nella malattia metabolica. Come tale, l’inibizione di AMYR o TRVP4 in certi momenti della malattia metabolica e nelle prime fasi del diabete può essere garantita per bloccare gli effetti tossici dell’amilina oligomerica o di Aβ. Tuttavia, una forte evidenza suggerisce anche che la sostituzione dell’amilina con amilina umana o pramlintide riduce la maggior parte della patologia principale legata all’AD, migliorando anche la cognizione nei modelli di roditori di AD. Come tale, la sostituzione di segnalazione amilina con amilina o pramlintide nelle fasi medie e tardive del diabete, quando la segnalazione amilina è perso a causa di aggregazione, oligomerizzazione, o la perdita delle cellule β può essere garantita. A tal fine, c’è anche la necessità di discernere la presentazione temporale degli eventi patologici nell’invecchiamento del cervello metabolicamente collegato e le opzioni terapeutiche per la malattia in fase iniziale, intermedia e tardiva. L’analisi critica e la verifica della natura diretta e delle capacità di segnalazione di questi amiloidi così come la natura terapeutica dei trattamenti temporali specifici possono aiutare a colmare il divario tra le terapie di inibizione AMYR e le terapie di sostituzione dell’amilina.
Finanziamento
Il finanziamento di questo articolo è stato fornito dal National Institutes of Aging grant 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau e disfunzione sinaptica. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. malattia di Alzheimer negli Stati Uniti (2010-2050) stimato utilizzando il censimento 2010. Neurologia. 2013; 80: 1778-1783.
- Associazione As. 2016 Alzheimer fatti e cifre della malattia di Alzheimer. Alzheimer & Demenza. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: pochi candidati, frequenti fallimenti. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Invecchiamento. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil e memantina per la malattia di Alzheimer da moderata a grave. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. livelli di glucosio e rischio di demenza. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesità e demenza: metanalisi e previsione corretta della prevalenza di demenza negli Stati Uniti e in Cina. Obesità. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabete mellito e rischio di demenza: una meta?analisi di studi prospettici osservazionali. Giornale di indagine del diabete. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesità nella mezza età e rischio futuro di demenza: uno studio longitudinale di 27 anni basato sulla popolazione. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Il diabete di tipo 2 come fattore di rischio per la malattia di Alzheimer: i confonditori, le interazioni e la neuropatologia associati a questo rapporto. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelera invecchiamento strutturale del cervello e declino cognitivo. Neurologia. 2011; 77: 461-468.
- Ginter E, Simko V. Prevalenza globale e futuro del diabete mellito In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. L’obesità e le sue condizioni comorbide. Clin Cornerstone. 1999; 2: 17-31.
- Federazione ID. Atlante del diabete IDF. Bruxelles: Federazione Internazionale del Diabete. 2013.
- K Dash S. Deterioramento cognitivo e diabete. Recenti Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Rischio di demenza tra le persone con diabete mellito: uno studio di coorte basato sulla popolazione. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Demenza e declino cognitivo nel diabete di tipo 2 e fasi prediabetiche: verso interventi mirati. Il lancetta Diabete & endocrinologia. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabete mellito e malattia di Alzheimer: patologia condivisa e trattamento. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalenza di diabete e prediabete e loro fattori di rischio tra gli adulti del Bangladesh: un sondaggio nazionale. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabete mellito e il rischio di demenza The Rotterdam Study. Neurologia. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzima regola i livelli di insulina, amiloide β-proteina, e la proteina β-amiloide precursore intracellulare dominio in vivo. Atti della National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulina legame ai capillari del cervello è ridotto in geneticamente obesi, ratti Zucker iperinsulinemici. Peptidi. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels increase During Intravenous Insulin Infusions in Man*. Il Journal of Clinical Endocrinology & Metabolismo. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. I livelli di insulina sono diminuiti nel liquido cerebrospinale di donne con malattia di Alzheimer prodomica. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Bersaglio multiplo di hAmylin sui neuroni ippocampali primari di ratto. Neuropharmacology. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Meccanismi che collegano l’obesità alla resistenza all’insulina e al diabete di tipo 2. Natura. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. alterata tolleranza al glucosio è associato con un aumento dell’immunoreattività del polipeptide amiloide dell’isolotto (IAPP) nelle cellule beta pancreatiche. Il giornale americano di patologia. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, e diabete mellito. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. L’attivazione dell’inflammasoma NLRP3 da polipeptide amiloide dell’isolotto fornisce un meccanismo per IL-1β aumentato nel diabete di tipo 2. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. marcatori infiammatori e rischio di diabete di tipo 2: una revisione sistematica e meta-analisi. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. β-amiloide di Alzheimer, amilina umana dell’isolotto e frammento di proteina prionica evocano aumenti di calcio libero intracellulare da un meccanismo comune in una linea cellulare ipotalamica GnRH neuronale. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Deposizione intraneuronale di amilina, lesione perossidativa della membrana e aumento della sintesi di IL-1β nel cervello di pazienti con malattia di Alzheimer con diabete di tipo 2 e in ratti HIP diabetici. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Deposito di amilina nel cervello: una seconda amiloide nella malattia di Alzheimer. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding e cross-seeding di amiloidosi localizzata: un legame molecolare tra diabete di tipo 2 e malattia di Alzheimer. La rivista americana di patologia. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Recettore dell’amilina: un obiettivo patofisiologico comune nella malattia di Alzheimer e nel diabete mellito. Fronte Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Il peptide amiloide β (Aβ) attiva direttamente il sottotipo di recettore amilina-3 innescando molteplici vie di segnalazione intracellulari. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide Antagonizes Beta Amyloid (Aβ)-and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. La depressione indotta dalla beta amiloide del potenziamento a lungo termine ippocampale è mediata attraverso il recettore dell’amilina. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Iniezione intraperitoneale del peptide pancreatico amilina riduce potentemente la compromissione comportamentale e la patologia amiloide del cervello in modelli murini della malattia di Alzheimer. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. I ligandi del recettore dell’amilina riducono la cascata patologica della malattia di Alzheimer. Neuropharmacology. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilizzazione di β-catenina da mutazioni in presenilina-1 potenzia l’apoptosi neuronale. Natura. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetato injection for the treatment of type 1 and type 2 diabetes mellitus. Clin Ther. 2007; 29: 535-562.
- Singh?Franco D, Perez A, Harrington C. L’effetto di pramlintide acetato sul controllo glicemico e il peso in pazienti con diabete mellito di tipo 2 e in pazienti obesi senza diabete: una revisione sistematica e meta’analisi. Diabete, obesità e metabolismo. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide come aggiunta alla terapia insulinica migliora il controllo glicemico e del peso a lungo termine in pazienti con diabete di tipo 2. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. La rivista americana di patologia. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. AC253 ciclico, un nuovo antagonista del recettore dell’amilina, migliora i deficit cognitivi in un modello murino della malattia di Alzheimer. Alzheimer & Demenza: Translational Research & Interventi clinici. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Ridotto comportamento nocicettivo in topi knockout del polipeptide amiloide delle isole (amilina). Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Attività di pramlintide, ratto e amilina umana ma non Aβ1-42 ai recettori amilinici umani. Endocrinologia. 2014; 155: 21-26.