John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstrakt
Stále více důkazů poukazuje na úzký vztah mezi diabetem II. typu (T2D) a Alzheimerovou chorobou (AD). Důležité je, že tato dvě onemocnění mají řadu patologických podobností, včetně akumulace amyloidu, oxidačního stresu, zánětu a buněčné smrti. Dosud chybějí léky pro AD a T2D a existuje zásadní potřeba objevit a vyvinout nová terapeutika pro tato onemocnění. Řada studií na lidech a hlodavcích poskytla důkazy o tom, že suplementace metabolickými hormony je velmi cenná pro zlepšení kognitivních funkcí a celkového metabolického zdraví u T2D i AD. Hormon slinivky břišní amylin se objevil jako klíčová součást etiologie onemocnění T2D i AD, ačkoli přesná role, kterou amylin u těchto onemocnění hraje, není dosud dobře známa. V tomto článku kriticky hodnotíme současnou literaturu, která využívá lidský amylin nebo jeho syntetický analog, pramlintid, a také antagonisty amylinových receptorů pro léčbu AD.
Úvod
Alzheimerova choroba (AD) je progresivní, vysilující neurodegenerativní onemocnění charakterizované hromaděním plaků amyloidu beta (Aβ) a neurofibrilárních spletí složených z hyperfosforylovaného tau1. Hromadění těchto patologických peptidů přispívá k deficitu výkonných funkcí, jako je učení a paměť, nálada, afekt atd., a představuje značnou zátěž pro pacienta i pečovatele. Výskyt Alzheimerovy choroby v USA narůstá alarmujícím tempem: odhaduje se, že v roce 2017 žilo 5,5 milionu Američanů s Alzheimerovou chorobou, a očekává se, že do roku 2050 se tento počet ztrojnásobí2. Kromě toho náklady na péči o pacienty s AD a jejich léčbu v současnosti přesahují 200 miliard dolarů ročně a očekává se, že se budou jen zvyšovat3. Ačkoli je AD v USA i mimo ně zjevně monumentálním problémem, možnosti léčby jsou stále velmi omezené4. Bylo provedeno mnoho zkoušek léků s širokou škálou cílených přístupů, avšak v současné době existuje pouze šest léků schválených úřadem FDA pro AD a jedná se pouze o symptomatickou léčbu5, 6. Většina dosud vyvinutých farmakologických látek se zaměřuje specificky na charakteristický znak patologie Aβ nebo tau, žádná z nich však nebyla úspěšná při odstraňování nebo prevenci patologie4. Proto je zásadní potřeba vyvinout životaschopná terapeutika a preventivní léčbu AD.
Věkem podmíněná (sporadická) AD je komplikované multifaktoriální onemocnění, které má četné genetické a environmentální vlivy. Na vzniku sporadické AD se významně podílí životní prostředí a životní styl; na vzniku AD se podílejí faktory jako strava7-9, obezita8-10, metabolický syndrom7, diabetes II. typu (T2D)9, 11 a kardiovaskulární onemocnění12. Zásadní význam má skutečnost, že míra obezity a diabetu rychle roste souběžně s výskytem AD12, 13. Ačkoli je vztah mezi obezitou a AD poněkud nejasný, existují důkazy, že obezita ve středním věku hraje roli při rozvoji AD10. Ještě důležitější je, že obezita je běžně doprovázena řadou dalších onemocnění, včetně kardiovaskulárních chorob, hypertenze, dyslipidemie, T2D, mozkové mrtvice atd.14 . Výskyt T2D rychle stoupá, podle odhadů CDC má v USA diabetes přibližně 30,3 milionu lidí (1 z 10 dospělých) a ohromujících 84,1 milionu (1 ze 3 dospělých) má prediabetes, přičemž většina z nich o svém onemocnění neví. Navíc se předpokládá, že v důsledku rozsáhlého poklesu fyzické aktivity, který je doprovázen současným nárůstem příjmu potravy a špatným stravováním, se počet případů obezity, T2D, metabolického syndromu a kardiovaskulárních onemocnění do roku 2035 celosvětově zvýší pouze na odhadovaných 600 milionů případů T2D15.
Soubor důkazů, podle nichž se metabolické funkce a onemocnění podílejí na procesu úbytku kognitivních funkcí a stárnutí, je značný16, 17 . Například přibližně 70 % osob s diagnózou T2D uvádí zhoršení kognitivních funkcí a u značného počtu pacientů s T2D se později vyvine demence16, 18-21. Jedinci s diagnózou T2D po dobu nejméně pěti let mají významně zvýšené riziko vzniku AD ve srovnání s těmi, kteří trpí T2D méně než pět let17. Tyto údaje společně naznačují, že rostoucí prevalence T2D v populaci může přispívat k rostoucímu výskytu AD.
T2D je zpočátku charakterizováno vysokou hladinou glukózy a inzulinu v krvi, což vede k hyperinzulinemii; důležité je, že amylin, malý metabolický hormon produkovaný buňkami β-isletů slinivky břišní, je společně s inzulinem balen a vylučován, a proto je u T2D nadprodukován22. Důležité je, že existuje řada patologických rysů, které jsou přítomny jak u T2D, tak u AD: 1) snížený metabolismus mozku a rezistence na metabolické hormony 2) patologie amyloidu 3) oxidační stres (OS) a zánět. Chronická, hyperinzulinemie a hyperamylinemie vede k řadě fyziologických problémů: chronická hyperinzulinemie vede k systémové inzulinové rezistenci22, zhoršenému transportu inzulinu přes hematoencefalickou bariéru (BBB)23, 24, a tím ke snížené inzulinové signalizaci v mozku25. Ztráta inzulinové signalizace v mozku je spojena s řadou patologických rysů souvisejících s AD, včetně zvýšené produkce Aβ, fosforylace tau a neurozánětu.
Kromě toho má amylin při vysokých koncentracích podobné patologické rysy jako Aβ26 a může být společnou cestou mezi oběma chorobami. Například fibrily amylinu byly nalezeny ve slinivce břišní u 95 % pacientů s T2D27-29 a způsobují řadu fyziologických poruch včetně aberantního influxu Ca2+, zvýšené sekrece prozánětlivých cytokinů30,31 a nakonec i ztrátu buněk β-isletů32. Amylin navíc snadno prochází přes BBB a vytváří amylinové fibrily i smíšené plaky s Aβ v mozku a může být zodpovědný za patologii podobnou AD a výsev Aβ u T2D33-35. Je známo, že amylin ovlivňuje dlouhodobou potenciaci (LTP) v hipokampu a může mít vrozený vliv na kognitivní funkce v mozku36-39 . Zda je amylin u těchto onemocnění toxickým inzultem, nebo zda jeho funkční ztráta v důsledku agregace nebo ztráty β-buněk v pozdním stadiu T2D přispívá k rozvoji AD, však zůstává nejasné.
Dichotomie amylinové signalizace
O zapojení amylinového receptoru (AMYR) a amylinové signalizace do progrese onemocnění a etiologie T2D a AD se stále hodně diskutuje. Soubor výzkumů zaměřených na rozeznání tohoto vztahu se rychle rozšiřuje. Všechny relevantní výzkumy důsledně prokazují, že modulace amylinové signalizace ovlivňuje patologii související s AD. Povaha tohoto vztahu však dosud nebyla konkrétně objasněna. Několik skupin přineslo přesvědčivé údaje, které naznačují, že amylinová signalizace je prospěšná při prevenci patologie související s AD a kognitivních deficitů jak in vivo, tak in vitro40-44 . Důležité je, že pramlintid, rekombinantní neagregační forma amylinu, který se používá ve spojení s inzulinovou terapií k léčbě diabetu a zlepšuje kontrolu glykémie, snižuje tělesnou hmotnost a snižuje sérové markery OS45-47 , se rovněž jeví jako slibný terapeutický prostředek pro AD. Dosud však neproběhly žádné klinické studie, které by byly zaměřeny na využití amylinu nebo pramlintidu jako terapeutického prostředku při léčbě demence. Jasné důkazy ze studií na hlodavcích naznačují, že chronická léčba lidským amylinem nebo pramlintidem představuje silný terapeutický přínos při snižování patologie související s AD; suplementace amylinem/pramlintidem snižuje hladiny rozpustného Aβ, zátěž plaku, fosforylaci tau, neurozánět a OS a zároveň zlepšuje kognici40-42,44 . Výše uvedené údaje naznačují, že ztráta vrozené signalizace amylinu v CNS v důsledku agregace dává vzniknout zvýšenému riziku rozvoje AD a podrobněji se jim věnuje Grizzanti et al. 201848.
Naopak studie také ukazují, že lidský amylin a Aβ mají podobné toxické účinky a že tyto toxické účinky lze zmírnit pomocí antagonisty AMYR36-39,49. Údaje například ukazují, že léčba antagonisty AMYR in vivo přináší velmi podobné fyziologické výhody jako léčba amylinem nebo pramlintidem. Léčba myší TgCRND8 s AD pomocí AC253, antagonisty AMYR, nebo jeho cyklického protějšku cAC253 snižuje neurozánět, hladiny rozpustného Aβ a zátěž plaku a zároveň zlepšuje kognici50. Podobně studie in vitro/ex-vivo ukazují, že nízké dávky lidského amylinu nebo Aβ způsobují narušení LTP a že tyto deficity jsou blokovány AC253 nebo pramlintidem38,39 a vyšší dávky lidského amylinu/amylinových oligomerů jsou spojeny s nekontrolovaným influxem Ca2+, který je silně spojen s buněčnou smrtí26,32 . Tyto údaje společně podporují toxickou funkci amylinových oligomerů, a tedy potenciální terapeutický mechanismus blokády AMYR. Jiní naopak prokázali, že příznivé účinky amylinu lze blokovat pomocí AC25341. Terapeutický potenciál léčby nebo inhibice amylinu tak zůstává nejasný a poukazuje na komplexní a dichotomickou povahu amyloidů v mozku a na periferii.
Skládání skládačky dohromady
V současné literatuře existuje řada děr, které je třeba zaplnit, aby byl obraz o amylinu úplnější: 1) povaha vrozeného amylinového systému a amylinové signalizace v mozku 2) možnosti signalizace Aβ a pramlintidu prostřednictvím tří hlavních AMYR a příbuzných receptorů 3) terapeutické mechanismy, kterými je zprostředkována inhibice amylinu/pramlintidu nebo AMYR. Za prvé, zajímavé nové údaje ukazují, že AMYR se podílí nejen na signalizaci, ale také na transportu ligandu přes BBB. AMYR je heterodimerní receptor, který se skládá z kalcitoninového receptoru a proteinu modifikujícího aktivitu receptoru (1-3)51 . Za tímto účelem 50 % globální knockdown kalcitoninového receptoru (klíčová součást AMYR) významně snížil množství AC253 nalezeného v mozku50 , což naznačuje, že AMYR umístěné v BBB se podílejí na transportu těchto ligandů do mozku a mohou se také podílet na přenosu amylinu a pramlintidu do/z mozku. Existence těchto transportních mechanismů v BBB naznačuje, že amylin má pravděpodobně vrozenou fyziologickou funkci v mozku, protože jeho transport do mozku je přísně kontrolován. Stále však není jasné, jak signalizace amylinu nebo její nedostatek vede k patologickým rysům AD a zda je AMYR prostředníkem, kterým Aβ zprostředkovává své toxické účinky.
Dále existují rozporuplné důkazy, pokud jde o vztah mezi Aβ a AMYR. Ačkoli několik studií jasně ukazuje, že lidský amylin a Aβ mají podobné účinky na LTP v CNS a použití inhibitorů AMYR tyto škodlivé účinky zmírňuje36-39, jiné důkazy naznačují, že Aβ (1-42) není schopen signalizovat přes jakoukoli AMYR, aby vyvolal jakoukoli odpověď cAMP při nejrůznějších koncentracích52. Je možné, že Aβ aktivuje různé signalizační kaskády prostřednictvím interakce s AMYR nebo prostě působí jako inertní kompetitivní inhibitor, ale to zatím nebylo prokázáno.
Samostatná studie navíc prokázala, že oligomerní amylin zprostředkovává své toxické účinky přímo prostřednictvím AMYR a nepřímo prostřednictvím TRPV4, neselektivního kationtového kanálu26. Nízké koncentrace až lidského amylinu vyvolávají odpověď Ca2+, která je zprostředkována jeho nativním receptorem. Při vyšších koncentracích však lidský amylin tvoří oligomery a aktivuje aberantní signalizaci, která vede k aktivaci kanálů TRVP4 a umožňuje nekontrolovaný přítok kationtů, zejména Ca2+. Farmakologická blokáda AMYR a TRPV4 ukazuje, že oba receptory jsou nezbytné k tomu, aby oligomerní lidský amylin vyvolal své toxické účinky na Ca2+26. Je tedy pravděpodobné, že Aβ zprostředkovává své toxické účinky na AMYR podobným způsobem, ačkoli tyto údaje zatím neexistují. Nekontrolovaný influx Ca2+ je spojen s řadou patologických jevů, včetně nekontrolovaného uvolňování vezikul, dysfunkce OS a mitochondrií, apoptózy atd. Za tímto účelem je pravděpodobné, že buněčná dysfunkce a rozvoj další patologie podobné AD, která vzniká v důsledku toxické amyloidové signalizace, je zprostředkována prostřednictvím AMYR i TRPV4. Proto je nutné rozlišit signální kaskády, které modulují vztah mezi AMYR a TRVP4. Dále jsou opodstatněné farmakologické experimenty, které využívají Aβ a pramlintid v široké škále dávek, aby se určily účinky Aβ a pramlintidu na Ca2+ proudy, LTP, produkci cAMP a další signální kaskády, aby se určily jejich signální schopnosti. Tyto experimenty pomohou zaplnit některé mezery v současné literatuře, pokud jde o AMYR a její zapojení do chorobných stavů (obr. 1).
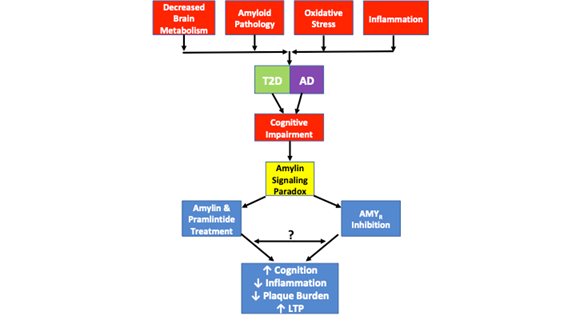
Obrázek 1. znázorňuje amylinový signální paradox a patologické podobnosti pozorované u T2D a AD. Snížený metabolismus mozku, patologie amyloidu, oxidativní stres a zánět jsou společné patologické rysy pozorované u obou onemocnění. I když ne každý případ T2D nebo AD zahrnuje každý z těchto patologických rysů, každý případ vykazuje kognitivní poruchu. Do hry vstupuje amylinový signální paradox, neboť studie ukázaly, že jak inhibice AMYR, tak agonismus AMYR prostřednictvím léčby amylinem a pramlintidem vedou ke zlepšení kognice, snížení zánětu, snížení zátěže plaky a zvýšení LTP. Mechanismy signalizace řízené agonismem a antagonismem AMYR ještě nebyly zcela objasněny. Zatímco amylinová signalizace je tradičně spojována se signalizací cAMP a PKA, není jasné, zda jsou amylinem/pramlintidem aktivovány i jiné kaskády. Dále není jasné, zda antagonisté AMYR, oligomery amylinu nebo Aβ signalizují prostřednictvím AMYR nebo zda mezi všemi těmito ligandy AMYR existuje nějaká podobnost nebo vzájemné ovlivňování. Proto řada experimentů navržených v tomto přehledu pomůže dále objasnit skutečnou povahu AMYR.
Závěry
Současný nesoulad, pokud jde o úlohu amylinové signalizace v mozku, ukazuje zásadní potřebu dalšího objasnění zapojení amylinu do AD i T2D. U T2D je pravděpodobné, že v časných stadiích onemocnění amylin zaplavuje mozek, vytváří oligomery, vyvolává aberantní signalizaci prostřednictvím svého nativního receptoru a rekrutuje TRPV4 k vyvolání patologického influxu Ca2+, což vede k rozsáhlé neuronální dysfunkci, která se projevuje jako OS, nekontrolované uvolňování vezikul a interneuronální dysfunkce, zánět a následná buněčná smrt. Tento mechanismus může být zodpovědný za počáteční přechod od zdravého mozku ke stárnutí mozku při metabolickém onemocnění. Inhibice AMYR nebo TRVP4 v určitých časových bodech metabolického onemocnění a v počátečních stadiích diabetu tak může být opodstatněná k zablokování toxických účinků oligomerního amylinu nebo Aβ. Silné důkazy však také naznačují, že náhrada amylinu buď lidským amylinem, nebo pramlintidem snižuje většinu hlavních patologických jevů souvisejících s AD a zároveň zlepšuje kognici u hlodavčích modelů AD. Náhrada amylinové signalizace amylinem nebo pramlintidem ve středních až pozdních stadiích diabetu, kdy dochází ke ztrátě amylinové signalizace v důsledku agregace, oligomerizace nebo ztráty β-buněk, tak může být opodstatněná. Za tímto účelem je také třeba rozlišit časovou prezentaci patologických dějů v metabolickém stárnutí mozku a terapeutické možnosti pro časné, střední a pozdní stadium onemocnění. Kritická analýza a testování přímé povahy a signalizačních schopností těchto amyloidů, jakož i terapeutické povahy specifických časových léčebných postupů může pomoci překlenout propast mezi terapií inhibice AMYR a terapií náhrady amylinu.
Financování
Financování tohoto článku bylo zajištěno grantem National Institutes of Aging 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimerova choroba: Aβ, tau a synaptická dysfunkce. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimerova choroba ve Spojených státech (2010-2050) odhadnutá na základě sčítání lidu z roku 2010. Neurologie. 2013; 80: 1778-1783.
- Asociace As. 2016 Alzheimerova choroba fakta a čísla. Alzheimerova & demence. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glukóza a riziko demence. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of dementia prevalence in the United States and China. Obezita. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27-year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Diabetes 2. typu jako rizikový faktor Alzheimerovy choroby: matoucí faktory, interakce a neuropatologie související s tímto vztahem. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurologie. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Obezita a její komorbidní stavy. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF diabetes atlas. Brusel: Mezinárodní federace pro diabetes. 2013.
- K Dash S. Kognitivní poruchy a diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. The lancet Diabetes & endocrinology. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalence diabetu a prediabetu a jejich rizikových faktorů mezi dospělými Bangladéšany: celostátní průzkum. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurologie. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Enzym odbourávající inzulín reguluje hladiny inzulínu, amyloidního β-proteinu a intracelulární domény β-amyloidního prekurzoru in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Vazba inzulinu na mozkové kapiláry je snížena u geneticky obézních, hyperinzulinemických Zuckerových potkanů. Peptides. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Hladiny inzulinu jsou sníženy v mozkomíšním moku žen s prodělanou Alzheimerovou chorobou. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neuropharmacology. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mechanismy spojující obezitu s inzulinovou rezistencí a diabetem 2. typu. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. The American journal of pathology. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimerův β-amyloid, amylin lidských ostrůvků a fragment prionového proteinu vyvolávají zvýšení intracelulárního volného vápníku společným mechanismem v hypotalamické neuronální buněčné linii GnRH. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronální depozita amylinu, peroxidativní poškození membrán a zvýšená syntéza IL-1β v mozku pacientů s Alzheimerovou chorobou a diabetem 2. typu a u diabetických potkanů HIP. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. The American journal of pathology. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylinový receptor: společný patofyziologický cíl u Alzheimerovy choroby a diabetes mellitus. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ) peptid přímo aktivuje podtyp receptoru amylin-3 spuštěním mnoha intracelulárních signálních drah. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintid antagonizuje depresi hipokampální dlouhodobé potenciace vyvolanou beta amyloidem (Aβ) a lidským amylinem. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloidem indukovaná deprese hipokampální dlouhodobé potenciace je zprostředkována přes amylinový receptor. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Intraperitoneální injekce pankreatického peptidu amylinu účinně snižuje poruchy chování a patologii mozkového amyloidu u myších modelů Alzheimerovy choroby. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylin receptor ligands reduce the pathological cascade of Alzheimer’s disease. Neuropharmacology. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotektivní účinky amylinového analogu pramlintidu na patogenezi a kognici Alzheimerovy choroby. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilizace β-kateninu mutacemi v presenilinu-1 potencuje neuronální apoptózu. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetate injection for the treatment of type 1 and type 2 diabetes mellitus. Clin Ther. 2007; 29: 535-562.
- Singh?Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta?analysis. Diabetes, obezita a metabolismus. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintid jako doplněk léčby inzulinem zlepšuje dlouhodobou kontrolu glykémie a hmotnosti u pacientů s diabetem 2. typu. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyclic AC253, a novel amylin receptor antagonist, improves cognitive deficits in a mouse model of Alzheimer’s disease. Alzheimerova & demence: Dement: Translational Research & Clinical Interventions. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Aktivita pramlintidu, potkaního a lidského amylinu, ale ne Aβ1-42 na lidských amylinových receptorech. Endokrinologie. 2014; 155: 21-26.