Alexei Drummond, Andrew Rambaut, Remco Bouckaert, and Walter Xie
1 Úvod
Tento tutoriál představuje software BEAST pro Bayesovskou evoluční analýzu prostřednictvím jednoduchého tutoriálu. Tutoriál zahrnuje spoluodhad fylogeneze genů a souvisejících divergenčních časů za přítomnosti kalibračních informací z fosilních důkazů.
Potřebujete mít k dispozici následující software:
- BEAST – tento balíček obsahuje program BEAST, BEAUti, TreeAnnotator a další obslužné programy. Tento návod je napsán pro BEAST v2.2.x, který má podporu více oddílů. Je k dispozici ke stažení na adrese http://www.beast2.org/.
- Tracer – tento program slouží ke zkoumání výstupu programu BEAST (a dalších bayesovských MCMC programů). Graficky a kvantitativně shrnuje rozdělení spojitých parametrů a poskytuje diagnostické informace. V době psaní tohoto textu je aktuální verze v1.6. Je k dispozici ke stažení na adrese
http://tree.bio.ed.ac.uk/software/. - FigTree – jedná se o aplikaci pro zobrazování a tisk molekulárních fylogenezí, zejména těch získaných pomocíBEAST. V době psaní tohoto článku je aktuální verze v1.4.2. Je k dispozici ke stažení na adrese http://tree.bio.ed.ac.uk/software/.
Tento výukový program vás provede analýzou zarovnání sekvencí vybraných z dvanácti druhů primátů (viz obrázek 1). Cílem je odhadnout fylogenezi, rychlost evoluce na jednotlivých liniích a stáří nekalibrovanýchancestralních divergencí.
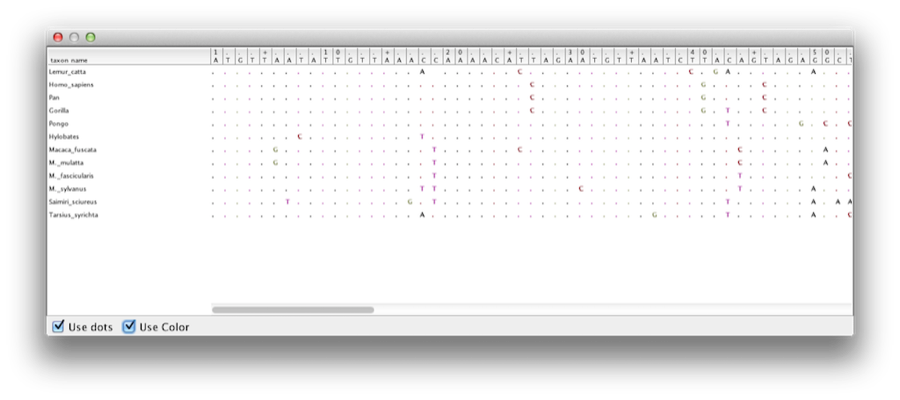
Obrázek 1: Část zarovnání pro primáty.
Prvním krokem bude převod souboru NEXUS s blokem DATA nebo CHARACTERS do vstupního souboru BEAST XML. To se provádí pomocí programu BEAUti (což je zkratka pro Bayesian Evolutionary Analysis Utility). Jedná se o uživatelsky přívětivý program pro nastavení evolučního modelu a možností pro analýzu MCMC. Druhým krokem je skutečné spuštění programu BEAST pomocí vstupního souboru vygenerovaného programem BEAUTi, kterýobsahuje data, model a nastavení analýzy. Posledním krokem je prozkoumání výstupu programu BEAST za účelem diagnostiky problémů a shrnutí výsledků.
2 BEAUti
Program BEAUti je uživatelsky přívětivý program pro nastavení parametrů modelu pro BEAST. Program BEAUti spustíte poklepáním na jeho ikonu. Po spuštění bude program BEAUti vypadat podobně bez ohledu na to, na jakém počítačovém systému je spuštěn. Pro tento návod je na obrázcích použita verze pro Mac OS X, ale verze pro Linux a Windows budou mít stejné uspořádání a funkce.
2.1 Načtení souboru NEXUS
Chcete-li načíst zarovnání ve formátu NEXUS, jednoduše vyberte možnost Importovat zarovnání… z nabídky Soubor nebo přetáhněte soubor doprostřed panelu Partitions.
Příkladový soubor s názvem primate-mtDNA.nex je k dispozici v adresáři examples/nexus/ pro Mac a Linux a examples/nexus/ pro Windows uvnitř adresáře, kam byl nainstalován BEAST. tento soubor obsahuje zarovnání sekvencí 12 druhů primátů.
Pokud je nainstalován příslušný balíček, zobrazí se okno Add Partition (Obrázek 2). Pokud používáte „čistý“ BEAST 2, můžete přejít k dalšímu odstavci. V opačném případě vyberte možnost Add Alignment (Přidat zarovnání) a pokračujte kliknutím na tlačítko OK.
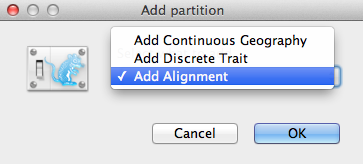
Obrázek 2: Okno Add Partition (Přidat oddíl) (zobrazí se pouze v případě, že jsou nainstalovány související balíčky).
Pokud se kódování v oddílech překrývá, zobrazí se okno s varovným hlášením (obrázek 3). Přečtěte si je a pokračujte kliknutím na tlačítko OK.
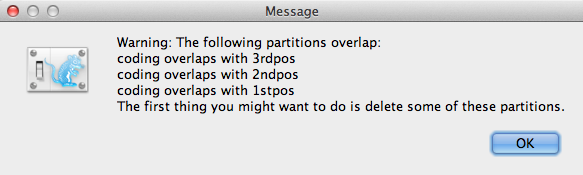
Obrázek 3: Okno s varovným hlášením (zobrazí se pouze v případě, že se v oddílech překrývá kódování).
Po načtení se na hlavním panelu zobrazí pět znakových oddílů (obrázek 4). Zarovnání je rozděleno na část kódující bílkoviny a nekódující část,přičemž kódující část je rozdělena na pozice kodonů 1, 2 a 3. Před pokračováním v dalším kroku musíte odstranit oddíl „coding“ (kódující), protože se vztahuje ke stejným nukleotidům jako oddíly „1stpos“, „2ndpos“ a „3rdpos“. Chcete-li odstranit oddíl „coding“, vyberte řádek a klikněte na tlačítko „-“ ve spodní části tabulky. Zarovnání můžete zobrazit dvojitým kliknutím na oddíl.
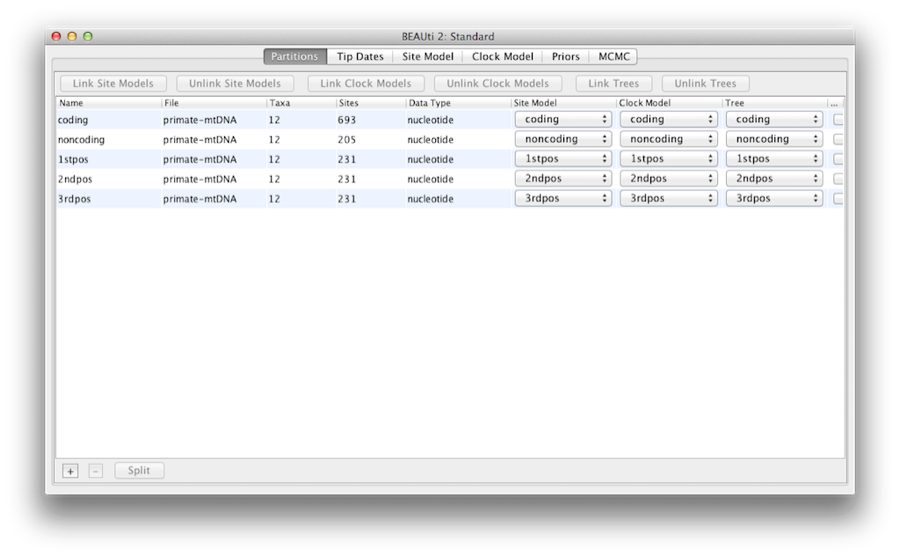
Obrázek 4: Snímek obrazovky karty dat v aplikaci BEAUti. Tento a všechny následující snímky obrazovky byly pořízeny na počítači Apple s operačním systémem Mac OS X a v jiných operačních systémech budou vypadat mírně odlišně.
Propojení/nepropojení modelů oddílů
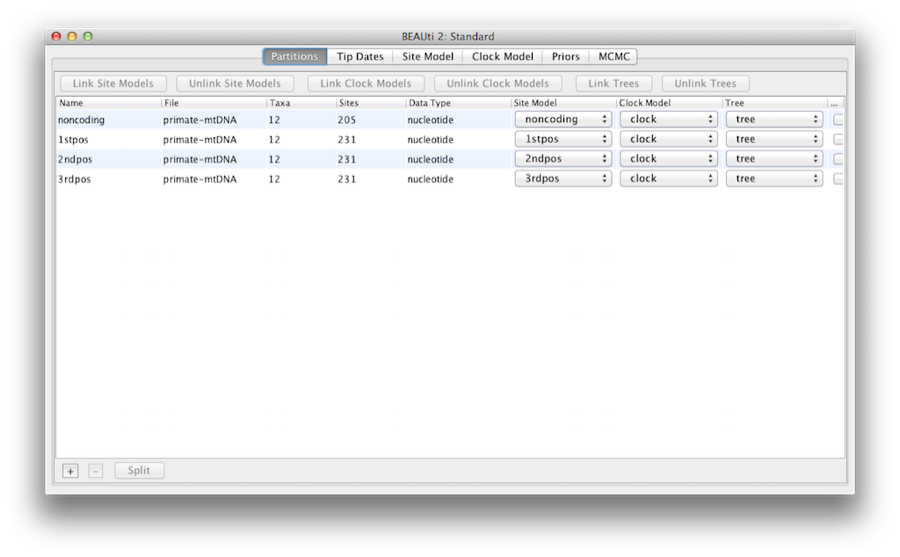
Obr. 5: Snímek obrazovky karty Oddíly v aplikaci BEAUti po propojení a přejmenování modelu a stromu hodin.
Protože jsou sekvence propojené (tj. všechny pocházejí z mitochondriálního genomu, o kterém se předpokládá, že u ptáků a savců nepodléhá rekombinaci), mají stejné předky, takže oddíly by měly sdílet stejný časový strom v modelu. Pro zjednodušení budeme také předpokládat, že oddíly sdílejí stejnou rychlost evoluce pro každou větev, a tedy i stejný „hodinový model“. omezíme naše modelování heterogenity rychlosti na heterogenitu mezi jednotlivými místy v každém oddílu a také připustíme, aby oddíly měly různé střední rychlosti evoluce.
V tomto bodě tedy budeme muset propojit hodinový model a strom. Na panelu Partitions vyberte v tabulce všechny čtyři oddíly (nebo žádný, ve výchozím nastavení se to týká všech oddílů) a klikněte na tlačítko Link Trees a poté na tlačítko Link Clock Models (viz obrázek 5). Poté klikněte na první rozevírací nabídku ve sloupci Clock Model a přejmenujte sdílený model hodin na „clock“. Stejně tak přejmenujte sdílený strom na „tree“. Tím se následující možnosti a generované soubory protokolu stanou přehlednějšími.
2.2 Nastavení substitučního modelu
Dalším krokem je nastavení substitučního modelu. Poté vyberte záložku Site Models v horní části hlavního okna (záložku Tip Dates přeskočíme, protože všechny taxony pocházejí ze současných vzorků). Tím se zobrazí nastavení evolučního modelu pro BEAST. Dostupné možnosti závisí na tom, zda se jedná o nukleotidy, nebo aminokyseliny, binární data, nebo obecná data. Nastavení, která se zobrazí po načtení zarovnání nukleotidů primátů, budou výchozí hodnoty pro nukleotidová data, takže musíme provést některé změny.
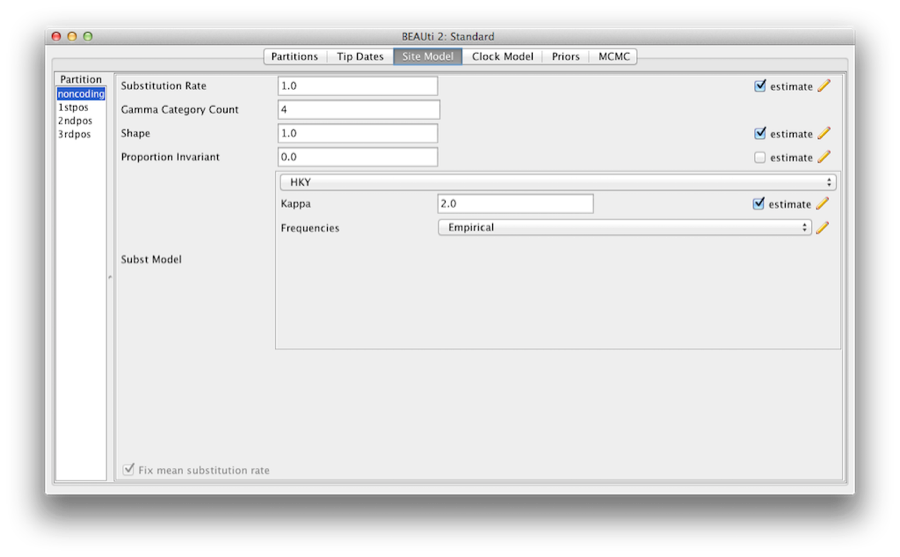
Obrázek 6: Snímek obrazovky karty modelu lokality v aplikaci BEAUti.
Většinu modelů byste měli znát. Nejprve nastavte hodnotu Gamma Category Count (Počet kategorií gama) na 4 a poté zaškrtněte políčko „odhad“ u parametru Shape (Tvar). To umožní modelovat rozdíly v míře mezi lokalitami v jednotlivých oddílech. Všimněte si, že 4 až 6 kategorií funguje dostatečně dobře pro většinu souborů dat, zatímco mít více kategorií vyžaduje více času na výpočet bez většího přínosu. Položku Proportion Invariant ponecháme nastavenou na nulu.
Poté z rozevírací nabídky Subst Model vyberte HKY. V ideálním případě by měl být vybrán substituční model, který nejlépe odpovídá údajům pro každý oddíl, ale zde pro zjednodušení použijeme HKY pro všechny oddíly. Dále z rozevírací nabídky Frequencies (Frekvence) vyberte Empirical (Empirické). Tím se frekvence zafixují na poměry pozorované v datech (pro každý oddíl zvlášť, jakmile odpojíme modely lokalit). Tento přístup znamená, že můžeme získat dobrou shodu s daty, aniž bychom tyto parametry explicitně odhadovali. Zde to děláme jednoduše proto, aby byly soubory protokolů o něco kratší a čitelnější v pozdějších částech cvičení.
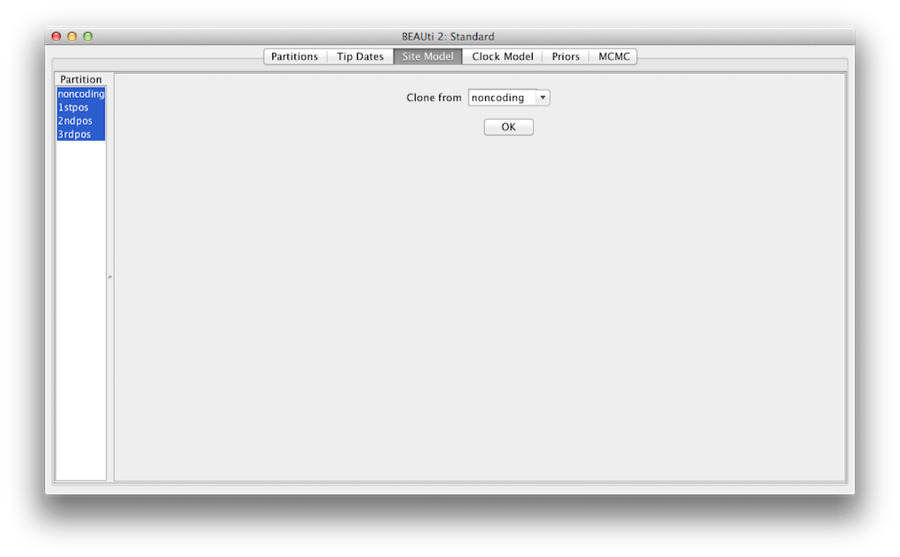
Obrázek 7: konfigurace klonování z jednoho modelu lokality do ostatních
Nakonec zaškrtněte políčko „odhadnout“ u parametru Substituční míra a zaškrtněte políčko Opravit průměrnou mutační míru. To umožní, aby se u jednotlivých oddílů odhadly relativní míry pro nepropojené modely lokalit (obrázek 6).
Nakonec podržte klávesu ‚shift‘, abyste vybrali všechny modely lokalit na levé straně, a kliknutím na tlačítko OK naklonujte nastavení z nekódujícího do 1.pos, 2.pos a 3.pos (obrázek 7). Projděte jednotlivé modely lokalit, jak vidíte, jejich konfigurace jsou nyní stejné.
2.3 Nastavení modelu hodin
Dalším krokem je výběr karty Modely hodin v horní části hlavního okna. Zde vybereme model molekulárních hodin. Pro toto cvičení ponecháme výběr na výchozí hodnotě striktních molekulárních hodin, protože tato data jsou velmi podobná hodinám a nepotřebují variaci rychlosti mezi větvemi, aby byla zahrnuta do modelu.
Chcete-li otestovat podobnost s hodinami, můžete (i) provést analýzu s modelem uvolněných hodin a zkontrolovat, jak velké rozdíly mezi rychlostmi jsou implikovány daty (více o tom viz variační koeficient), nebo(ii) provést porovnání modelu mezi striktními a uvolněnými hodinami pomocí pathsampling, nebo (iii) použít náhodný model lokálních hodin, který explicitně zvažuje, zda každá větev ve stromě potřebuje vlastní rychlost větvení.
2.4 Priory
Záložka Priory umožňuje zadat priory pro každý parametr v modelu. Výběry modelu provedené na kartách Model lokality a Model hodin vedou k zahrnutí různých parametrůdo modelu a ty jsou zobrazeny na kartě Priors (viz obrázek 8).
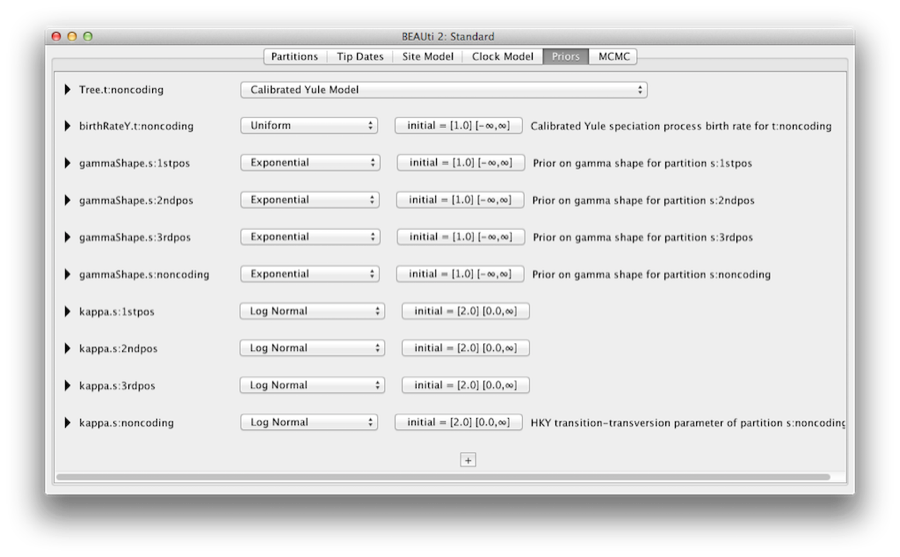
Obr. 8: Snímek obrazovky karty Priors v aplikaci BEAUti.
Tady také zadáme, že jako prioritu stromu chceme použít kalibrovaný model Yule. Yuleho model je jednoduchý model speciace, který je obecně vhodnější při uvažování sekvencí zrůzných druhů. Z rozbalovací nabídky Stromové priority vyberte Kalibrovaný Yuleho model.
2.4.1 Definování kalibračního uzlu
Nyní musíme zadat prioritu rozdělení kalibrovaného uzlu na základě našichznalostí. Tomu se říká kalibrace našeho stromu. Chcete-li definovat další prioritu, stiskněte malé tlačítko + pod seznamem priorit. Pokud není ve vašem zobrazení viditelné, posuňte panel až dolů, kde tlačítko + najdete. Zobrazí se dialogové okno, které vám umožní definovat podmnožinu taxonů ve fylogenetickém stromu. Jakmile vytvoříte sadu taxonů, budete moci později přidat kalibrační informace pro jejich nejnovějšího commonancestora (MRCA).
Pojmenujte sadu taxonů vyplněním položky označení sady taxonů. Nazvěte ji human-chimp, protože bude obsahovat taxony pro Homo sapiens a Pan. V následujícím seznamu uvidíte dostupné taxony. Postupně vyberte každý z obou taxonů a stiskněte tlačítko se šipkou > >. (Obrázek 9). klikněte na tlačítko OK a nově definovaná sada taxonů bude přidána do seznamu priorů. protože se jedná o kalibrovaný uzel, který se má používat ve spojení s kalibrovaným priorem Yule, musí být vynucena monofylie, proto zaškrtněte políčko označené Monophyletic. Tím omezíte topologii stromu tak, aby bylo seskupení člověk-šimpanz v průběhu analýzy MCMC zachováno jako monofyletické.
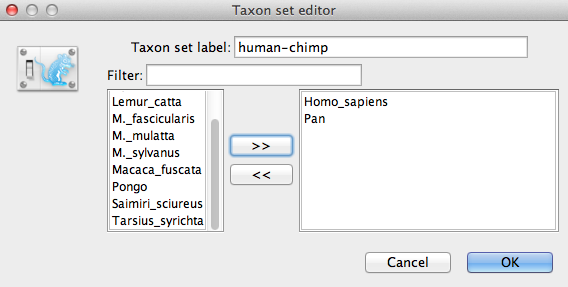
Obrázek 9: Editor sady taxonů v aplikaci BEAUti.
Pro zakódování kalibrační informace musíme zadat rozdělení pro MRCA člověka-šimpanze. z rozbalovací nabídky napravo od nově přidaného člověka-šimpanze.prior vyberte Log-normální rozdělení. Klikněte na černý trojúhelník a zobrazí se graf funkce hustoty pravděpodobnosti spolu s parametry logaritmicko-normálního rozdělení. nastavíme M=1,78 a S=0,085, což určí rozdělení se středem přibližně 6 milionů let se směrodatnou odchylkou přibližně 0,5 milionu let. Tím získáme střední 95% pravděpodobnostní rozsah pokrývající 5-7 Mya. To zhruba odpovídá současnému konsenzuálnímu odhadu data nejnovějšího společného předka lidí a šimpanzů (obrázek 10).
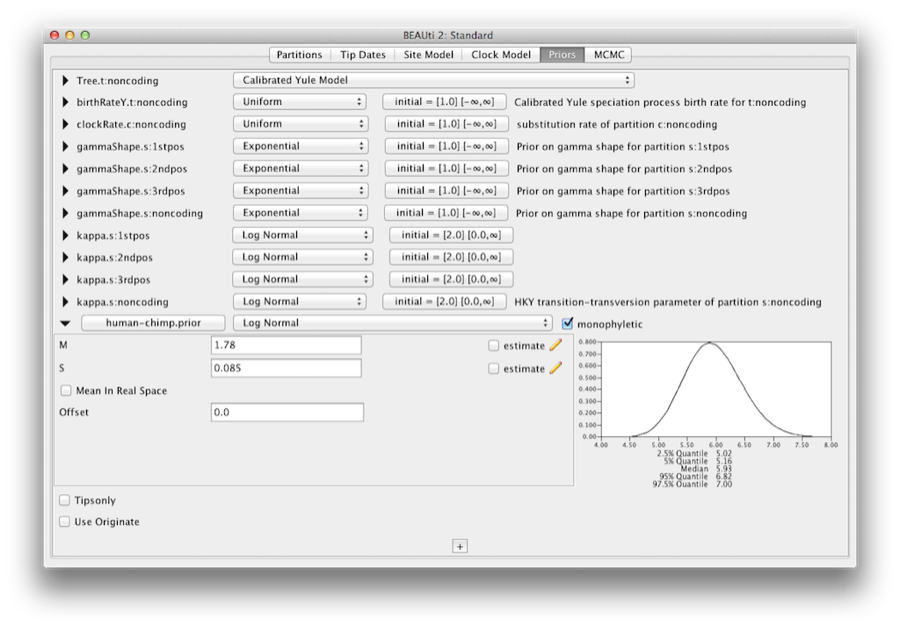
Obr. 10: Snímek obrazovky možností kalibrační priority v panelu Priors v aplikaci BEAUti.
Měli bychom se přesvědčit, že priory zobrazené v panelu priorů skutečně odrážejí předběžné informace, které máme o parametrech modelu. Nakonec také určíme některé rozptýlené „neinformativní“, ale správné priory na celkovou rychlost molekulárních hodin (clockRate) a rychlost speciace (birthRateY) priority Yuleova stromu. Pro každou z nich vybereme z rozbalovací nabídky možnost Gammaa pomocí tlačítka se šipkou rozbalíme zobrazení, abychom odhalili parametry priorityGamma. Jak pro hodinovou rychlost, tak pro míru porodnosti Yule nastavte parametr Alpha(shape) na 0,001 a parametr Beta (scale) na 1000 (obrázek 11).
Ve výchozím nastavení má každý z parametrů tvaru priority Gamma exponenciální rozdělení se střední hodnotou 1. To znamená (viz obrázek 3.7), že očekáváme určitou variabilitu. Ve výchozím nastavení mají parametry kappa pro model HKY lognormální(1,1,25) apriorní rozdělení, které se v podstatě shoduje s empirickými důkazy o rozsahu realistických hodnot pro přechod/přechod. Tyto výchozí priority jsou ponechány, protože jsou vhodné pro tuto konkrétníanalýzu.
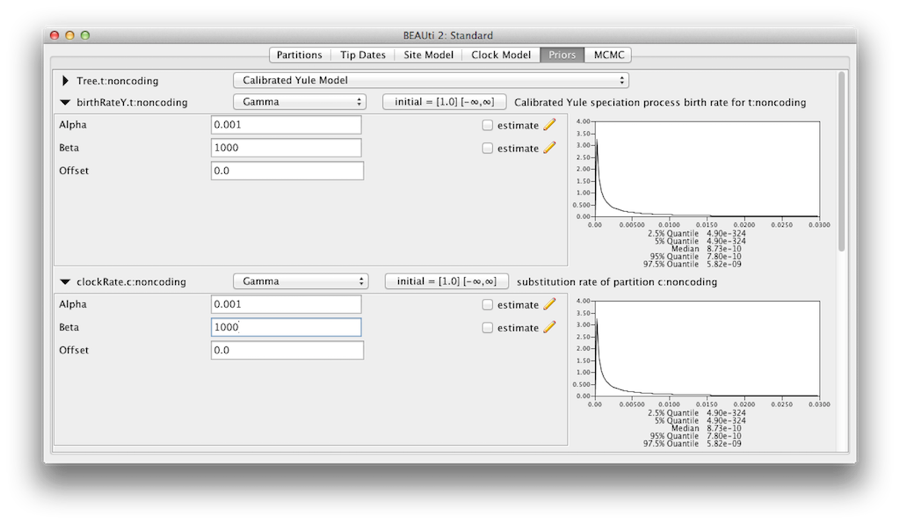
Obrázek 11: Gama priorita.
2.5 Nastavení možností MCMC
Další karta, MCMC, poskytuje obecnější nastavení pro kontrolu délky běhu MCMC a názvů souborů.
Nejprve máme k dispozici Délku řetězce. Jedná se o počet kroků, které MCMC provede v řetězci před ukončením. Jak dlouhý by měl být, závisí na velikosti datového souboru, složitosti modelu a požadované kvalitě odpovědi. Výchozí hodnota 10 000 000 je zcela libovolná a měla by být upravena podle velikosti vašeho souboru dat. Pro tuto datovou sadu nastavme délku řetězce na 6 000 000, protože na většině moderních počítačů to poběží poměrně rychle (několik minut).
Pole Store Every určuje, jak často se stav ukládá do souboru. Periodické ukládání stavu je užitečné v situacích, kdy výpočetní prostředí není příliš spolehlivé a běh BEASTu může být přerušen. Mít uloženou kopii posledního stavu umožňuje pokračovat v řetězci namísto opětovného spuštění od začátku, takže nemusíte znovu procházet vypalováním. pole Pre Burnin určuje počet vzorků, které nejsou zaznamenány na samém začátku analýzy. Pole Store Every a Pre Burnin ponecháme nastavena na výchozí hodnoty. Níže jsou uvedeny podrobnosti o souborech protokolu. Každý z nich lze rozbalit kliknutím na černý trojúhelník.
Další volby určují, jak často se mají hodnoty parametrů v Markovově řetězci zobrazovat na obrazovce a zaznamenávat do souboru protokolu.Výstup na obrazovku slouží pouze ke sledování průběhu programu a může být nastaven na libovolnou hodnotu (pokud je však nastaven na příliš malou hodnotu, množství informací zobrazovaných na obrazovce ve skutečnosti zpomaluje program). Pro soubor protokolu by měla být hodnota nastavena vzhledem k celkové délce řetězce. Příliš časté vzorkování bude mít za následek příliš velké soubory s malým dodatečným přínosem z hlediska přesnosti analýzy. Pokud budete vzorkovat příliš zřídka, soubor protokolu nebude zaznamenávat dostatečné informace o rozložení parametrů. Pravděpodobně se budete chtít zaměřit na ukládání ne více než 10 000 vzorků, takže by to nemělo být menší než délka řetězce / 10 000.
Pro toto cvičení nastavíme frekvenci logu sledování a logu stromů na 1 000 a logu obrazovky na 10 000. Zadejte také Primates.log jako název souboru logu sledování a Primates.trees jako název souboru logu stromů.Ujistěte se, že název souboru protokolu obrazovky zůstal prázdný, jinak se protokol obrazovky nebude zapisovat na obrazovku.
- Pokud používáte operační systém Windows, doporučujeme přidat k oběma těmto souborům příponu .txt (tedy Primates.log.txt a Primates.trees.txt), aby je systém Windows rozpoznal jako textové soubory.
2.6 Generování souboru BEAST XML
Nyní jsme připraveni vytvořit soubor BEAST XML. Za tímto účelem vyberte v nabídce Soubor možnost Uložit. Zaškrtněte výchozí priory a uložte soubor s vhodným názvem(obvykle končíme název souboru koncovkou .xml, tj. Primates.xml).nyní jsme připraveni spustit soubor pomocí programu BEAST.
3 Spuštění BEASTu
Obrázek 12: Snímek obrazovky BEASTu.
Nyní spusťte BEAST a až se zeptá na vstupní soubor, zadejte jako vstup nově vytvořený soubor XML. BEAST pak poběží, dokud nedokončívykazování informací na obrazovku. Skutečné soubory s výsledky se uloží na disk do stejného umístění jako váš vstupní soubor. Výstup na obrazovku bude vypadat asi takto:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Všimněte si, že na začátku je několik užitečných informací týkajících se zarovnání a toho, které pravděpodobnosti stromů jsou použity. Na začátku běhu jsou také uvedeny všechny citace důležité pro analýzu, které lze snadno zkopírovat do rukopisů informujících o analýze. Poté následuje hlášení o řetězci,které poskytuje určitou zpětnou vazbu v reálném čase o průběhu řetězce.
Na konci je vypsána analýza operátorů, která uvádí všechny operátory použité v analýze spolu s tím, jak často byl operátor vyzkoušen, přijat a odmítnut(viz sloupce #celkem, #přijat a #odmítnut). Míra přijetí je podíl případů, kdy je operátor přijat, když je vybrán pro provedení návrhu. Obecně platí, že vysoká míra přijetí, například nad 0,5, naznačuje, že návrhy jsou konzervativní a neprozkoumávají prostor parametrů efektivně. Na druhou stranu nízká míra přijetí naznačuje, že návrhy jsou příliš agresivní a téměř vždy vedou ke stavu, který je odmítnut kvůli nízkému posterioru. jak příliš vysoká, tak příliš nízká míra přijetí vede k nízkým hodnotám ESS. Pro mnoho (ale ne pro všechny) operátorů implementovaných v BEASTu je cílová míra přijetí 0,234 (na základě velmi omezených důkazů poskytnutých ).
Některé operátory mají parametr ladění, například faktor měřítka parametru ascale. Pokud se konečná míra přijetí neblíží cílové hodnotě, BEAST navrhne novou hodnotu ladicího parametru, která se vypíše v analýze operátoru. V tomto případě jsou všechny míry přijatelnosti dobré pro operátory, které mají ladicí parametry. Operátory bez parametrů ladění zahrnují pro tuto analýzu operátory wideexchange a Wilson-Balding. Oba tyto operátory se snaží měnit topologii stromu s velkými kroky, ale protože data v drtivé většině podporují jedinou topologii, jsou tyto radikální návrhy téměř vždy zamítnuty.
4 Analýza výsledků
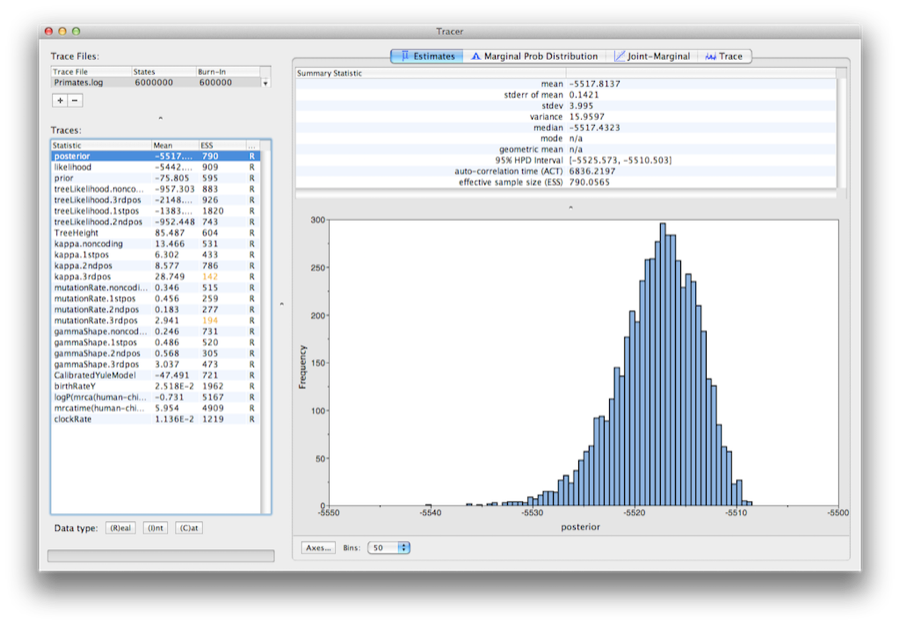
Obrázek 13: Snímek obrazovky programu Tracer v1.6.
Spustíte program s názvem Tracer a analyzujete výstupy programu BEAST. Po otevření hlavního okna zvolte v nabídce File (Soubor) položku Import Trace File… (Importovat sledovací soubor…) a vyberte soubor, který vytvořil program BEAST s názvem Primates.log (obrázek 13).
Pamatujte, že MCMC je stochastický algoritmus, takže skutečná čísla nebudou přesně stejná jako čísla zobrazená na obrázku.
Na levé straně je seznam různých veličin, které BEAST zaznamenal do souboru. Jsou zde stopy pro posterior (jedná se o přirozený logaritmus součinu pravděpodobnosti stromu a hustoty prior) a spojité parametry. Výběr stopy na levé straně vyvolá analýzy pro tuto stopu na pravé straně v závislosti na vybrané kartě. Při prvním otevření je vybrána „posteriorní“ stopa a v záložce Estimates (Odhady) jsou zobrazeny různé statistiky této stopy. v pravé horní části okna je tabulka vypočtených statistik pro vybranou stopu.
Zvolením parametru clockRate v levém seznamu se zobrazí průměrná rychlost vývoje (zprůměrovaná pro celý strom a všechna místa). Tracer vykreslí (marginální posteriorní) histogram pro vybranou statistiku a také vám poskytne souhrnné statistiky, jako je průměr a medián. Zkratka 95 % HPD znamená interval s nejvyšší posteriorní hustotou a představuje nejkompaktnější interval na vybraný parametr, který obsahuje 95 % posteriorní pravděpodobnosti. Volně si jej lze představit jako bayesovskou obdobu intervalu spolehlivosti. Parametr TreeHeight udává marginální posteriorní rozdělení stáří kořene celého stromu.
Zvolte parametr TreeHeight a poté klikněte klávesou Ctrl mrcatime(human-chimp) (v systému Mac OS X klikněte příkazem Command). Zobrazí se stáří kořene a kalibrační MRCA, které jsme dříve zadali v BEAUti. Můžete si ověřit, že divergence, kterou jsme použili ke kalibraci stromu(mrcatime(human-chimp)), má posteriorní rozdělení, které odpovídá námi zadanému apriornímu rozdělení (obrázek 14).
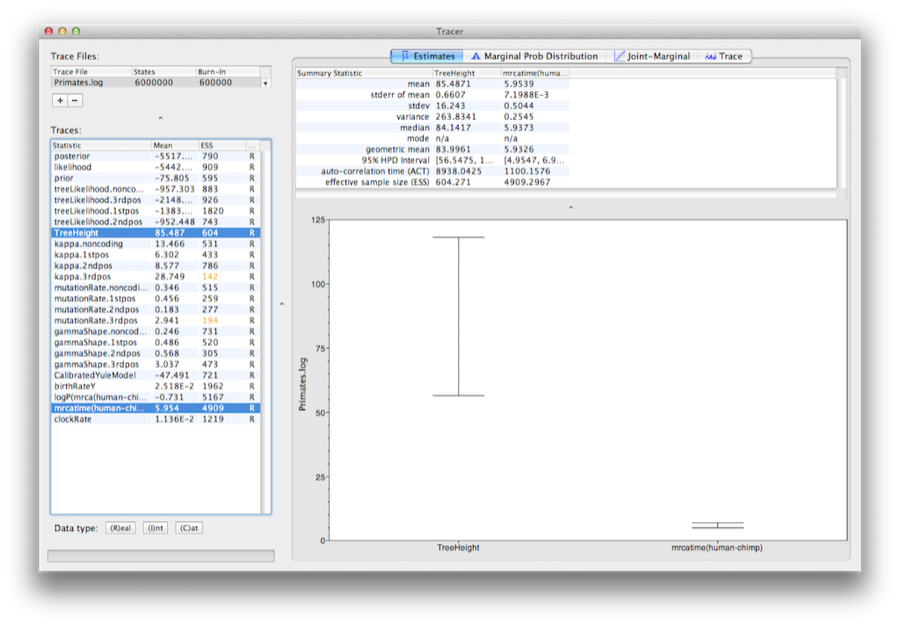
Obr. 14: Snímek obrazovky 95% HPD intervalů výšky kořene a uživatelem zadaného (human-chimp) MRCA v programu Tracer.
5 Marginální posteriorní odhady
Chcete-li zobrazit relativní míry pro čtyři oddíly, vyberte parametr mutationRate pro každý ze čtyř oddílů a vyberte záložku marginální hustoty v programu Tracer. obrázek 15 ukazuje marginální hustoty pro relativní míry substituce. Z grafu je patrné, že kodonové pozice 1 a 2 mají podstatně odlišné míry (0,456 versus 0,183) a obě jsou mnohem pomalejší než kodonová pozice 3 s relativní mírou2,941 . Nekódující oddíl má rychlost mezi kodonovými pozicemi1 a 2 (0,346). Dohromady tento výsledek naznačuje silnou purifikační selekci jak v kódujících, tak nekódujících oblastech zarovnání.
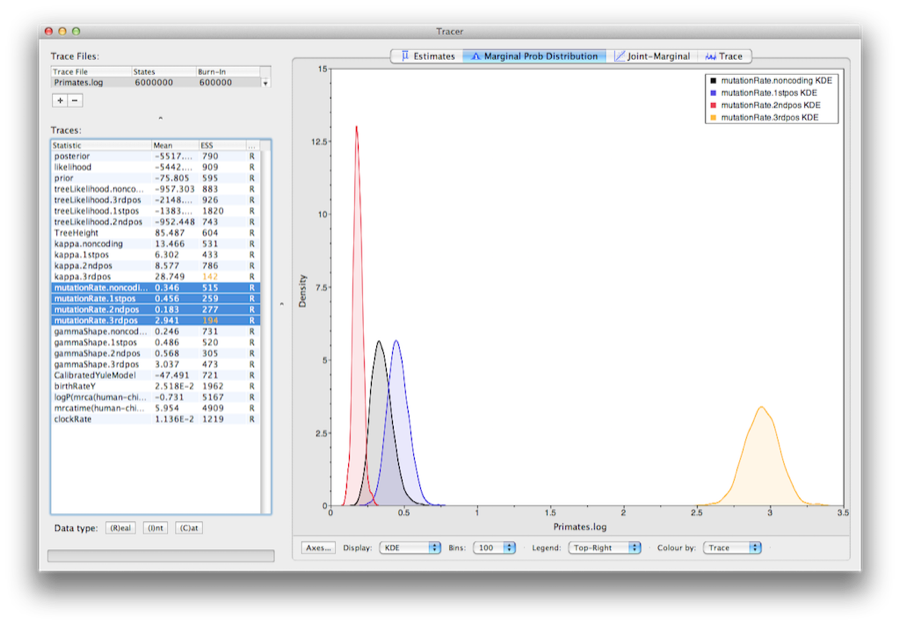
Obrázek 15: Snímek okrajových posteriorních hustot relativních substitučních rychlostí čtyř oddílů (vzhledem k průměrné rychlosti vážené místem).
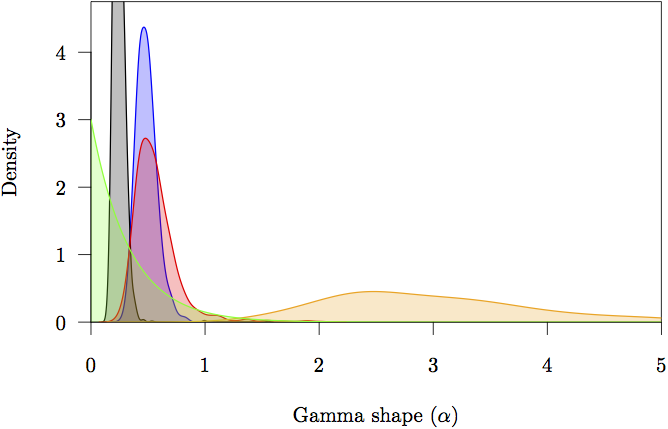
Obr. 16: Okrajové apriorní a posteriorní hustoty pro parametry tvaru (α). Priorita je vyznačena šedě. Zobrazen je také posteriorní odhad hustoty pro každé rozdělení: nekódující (oranžová) a první (červená), druhá (zelená) a třetí (modrá) pozice kodonu
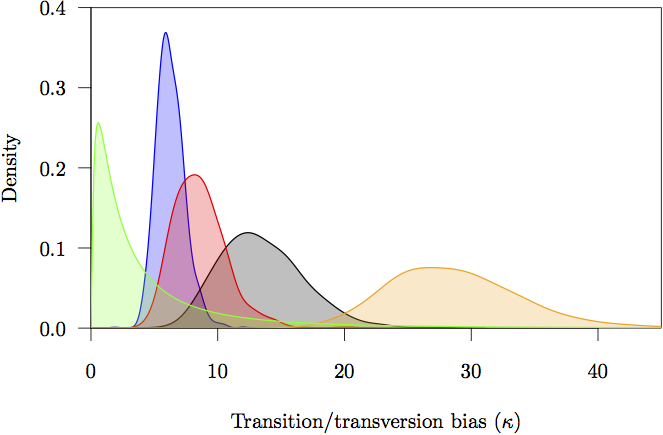
Obr. 17: Okrajová priorita a posteriorní hustota pro parametry zkreslení přechodu/tranverze (κ). Priorita je vyznačena šedě. Je také zobrazen odhad posteriorní hustoty pro každý oddíl: nekódující (oranžová) a pozice prvního (červená), druhého (zelená) a třetího (modrá) kodonu.
Otázky
Jaká je odhadovaná rychlost molekulární evoluce pro tento genový strom (uveďte 95% interval HPD)?
Jaké zdroje chyb tento odhad zahrnuje?
Jak starý je kořen stromu (uveďte průměr a 95% interval HPD)
6 Získání odhadu fylogenetického stromu
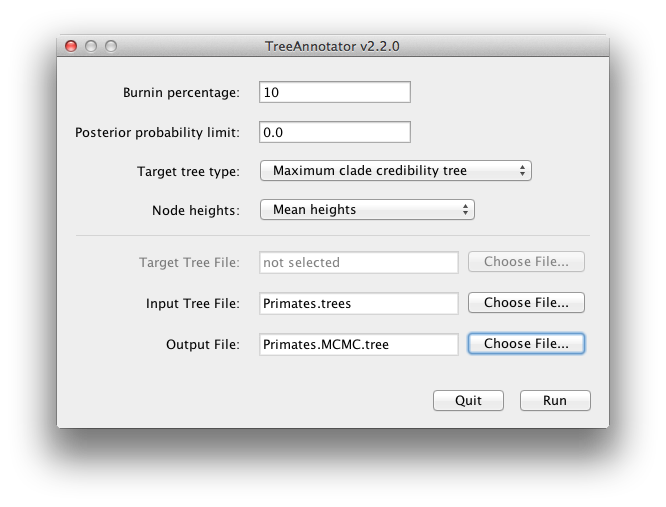
BEAST také vytváří posteriorní vzorek fylogenetických časových stromů spolu se svým vzorkem odhadů parametrů. Ty je třeba shrnout pomocí programu TreeAnnotator. Ten vezme soubor stromů a najde ten nejlépe podporovaný. Tento reprezentativní souhrnný strom pak anotuje průměrným stářím všech thenodů a odpovídajícími 95% rozsahy HPD. Vypočítá také posteriorní kladovou pravděpodobnost pro každý uzel. Spusťte program TreeAnnotator a nastavte jej podle obrázku 18.
Obrázek 18: Snímek obrazovky programu TreeAnnotator.
Vypálení je počet stromů, které se odstraní ze začátku vzorku. Na rozdíl od nástroje Tracer, který jako burnin zadává počet kroků, v nástroji TreeAnnotator je třeba zadat skutečný počet stromů. Pro tento běh jste zadali délku řetězce 6 000 000 kroků, který odebírá vzorky každých 1 000 kroků. Soubor stromů tedy bude obsahovat 6 000 stromů, a proto zadejte 10 % vyhořenív horním textovém poli.
Volba Limit posteriorní pravděpodobnosti určuje limit tak, že pokud se uzel ve vzorku stromů vyskytuje s menší četností než tento limit (tj. má posteriorní pravděpodobnost menší než tento limit), nebude anotován. Výchozí hodnota 0,5 znamená, že anotovány budou pouze uzly, které se vyskytují ve většině stromů. Pokud nastavíte tuto hodnotu na nulu, budou anotovány všechny uzly.
Typ cílového stromu určuje topologii stromu, která bude anotována. Můžete buď vybrat konkrétní strom ze souboru, nebo požádat TreeAnnotator, aby našel strom ve vašem vzorku. výchozí volba, Maximum clade credibility tree, najde strom s nejvyšším součinem posteriorní pravděpodobnosti všech jeho uzlů.
Pro výšky uzlů je výchozí volbou Common Ancestor Heights, která vypočítá výšku uzlu jako průměr času MRCA všech párů uzlů v klínu. U stromů s velkou nejistotou v topologii, a tedy mnoha kladů s nízkou podporou, mohou některé jiné metody vést ke stromům se zápornými délkami větví. V této analýze je podpora všech kladů v souhrnném stromu velmivysoká, takže to zde není problém. pro výšky uzlů zvolte střední výšky. Tím nastavíte výšky (stáří) každého uzlu ve stromu na průměrnou výšku v celém vzorku stromů pro daný klad.
Pro vstupní soubor vyberte soubor stromů, který vytvořil BEAST, a vyberte soubor pro výstup (zde jsme jej nazvali Primates.MCC.tree). Nyní stiskněte tlačítko Spustit a počkejte na dokončení programu.
7 Vizualizace odhadu stromu
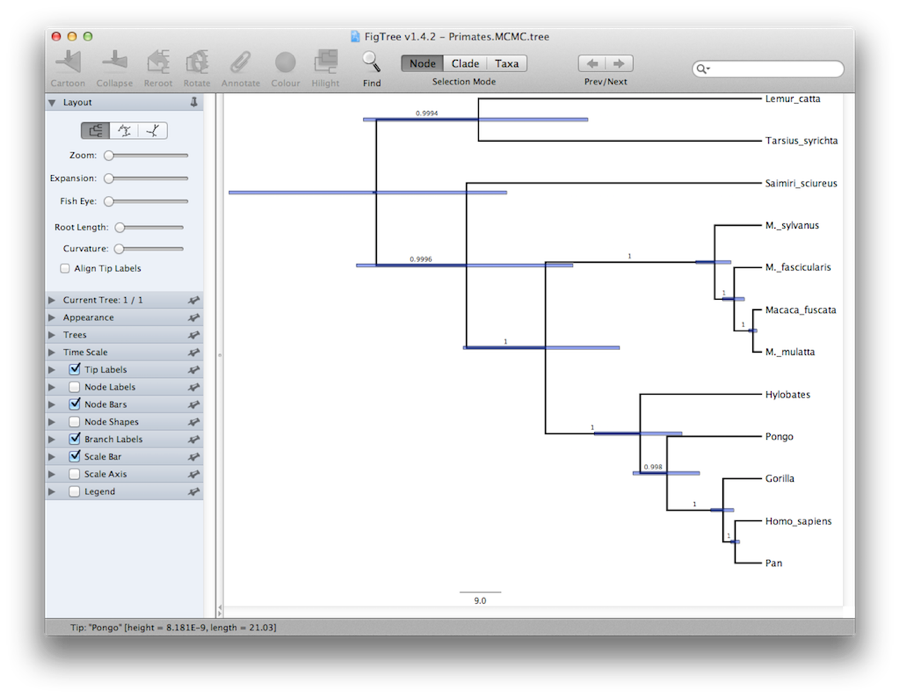
Nakonec můžeme strom vizualizovat v dalším programu nazvaném FigTree. Spusťte tento program a otevřete soubor Primates.MCC.tree pomocí příkazu Otevřít v nabídce Soubor. Strom by se měl zobrazit. nyní můžete zkusit vybrat některé z možností na ovládacím panelu vlevo. Nejprve v panelu expendujte možnost Trees a zaškrtněte Order nodes a zvolte Ordering by decreasing. Zkuste vybrat možnost Node Bars, abyste získali chybové sloupce stáří uzlů. Zapněte také možnost Branch Labels (Popisky větví) a vyberte možnost Posterior (Posterior), aby se pro každý uzel zobrazila posteriorní pravděpodobnost. Pokud používáte jiný než striktní hodinový model, pak v části Vzhled můžete programu FigTree také říci, aby větve obarvil podle míry. měli byste skončit s něčím podobným jako na obrázku 19.
Obrázek 19: Snímek obrazovky programů FigTree a DensiTree.
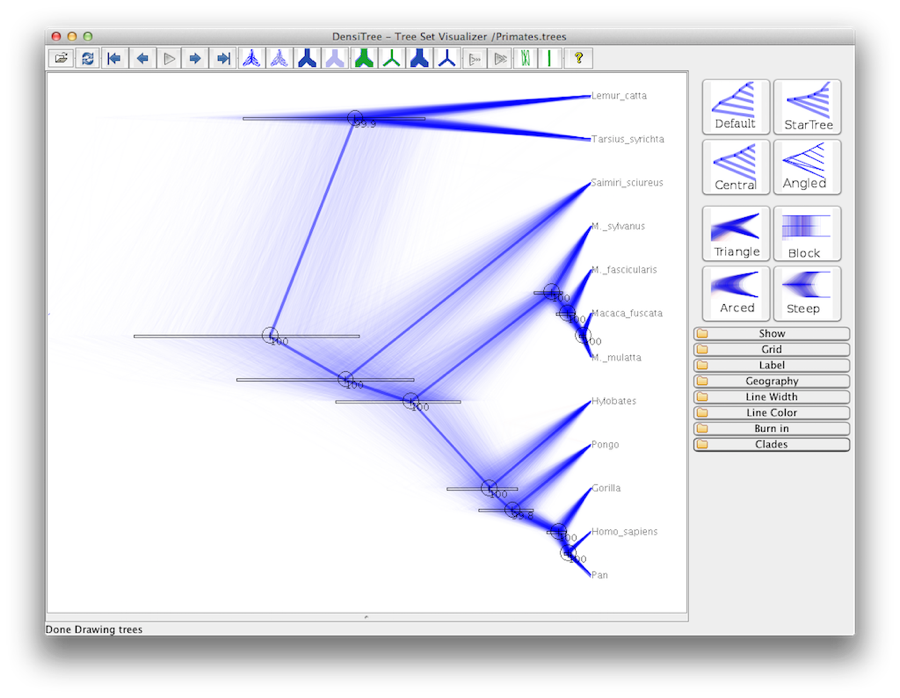
Alternativní zobrazení stromu lze provést pomocí programu DensiTree, který je součástí programu Beast 2. Výhodou programu DensiTree je, že dokáže vizualizovat jak nejistotu ve výškách uzlů, tak nejistotu v topologii. u této konkrétní sady dat je dominantní topologie přítomna ve více než 99 % vzorků. Došli jsme tedy k závěru, že výsledkem této analýzy je velmi vysoká shoda ohledně topologie (obrázek 19).
Otázky
- Rozlišuje se výrazně rychlost evoluce mezi jednotlivými liniemi ve stromu?
- DensiTree má panel kladů (Okno nabídky/Panel nástrojů Zobrazit klad), který zobrazuje informace o kladech.
Jaká je podpora kladů?
- V DensiTree můžete procházet topologie pomocí nabídky Procházet.Nejoblíbenější topologie má podporu více než 99 %.
Jaká je podpora druhé nejoblíbenější topologie?
- V nabídce nápovědy zobrazuje DensiTree některé informace.
Kolik topologií je v souboru stromu?
8 Porovnání výsledků s prioritou
Je dobré znovu provést analýzu při vzorkování z priority, abyste se ujistili, že interakce mezi prioritami neovlivňují informace o vaší prioritě. Interakce mezi priory může být problematická zejména při použití kalibrací, protože znamená vložení více priorů do stromu.
Pomocí BEAUti nastavte stejnou analýzu, ale v položce MCMC options vyberte možnost Sample from prior only. To vám umožní vizualizovat úplné rozdělení priorit při absenci vašich sekvenčních dat. Shrňte stromy z úplného priordistribuce a porovnejte souhrn s posteriorním souhrnným stromem.
Odhad doby divergence pomocí „datování uzlů“ typu popsaného v tétokapitole byl použit k zodpovězení řady různých otázek v ekologiia evoluci. Například datování uzlů pomocí fosilií bylo použito při určování druhové rozmanitosti cykasů , analýze rychlosti evoluce u kvetoucích rostlin a zkoumání původu sinic z teplých a studených pouští .
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong a další, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond a Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), č. 1, 114. A Gelman, G Roberts a W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled a Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and divergence time estimation, Syst Biol 61 (2012),no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little a S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), č. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian a Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molekulární biologie a evoluce 20 (2003), č. 6, 988-993. Stephen A Smith a Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008),no. 5898, 86-89.
Tento dokument byl přeložen z LATEXu pomocíHEVEA.