Úvod
Germline de novo mutace (DNM) jsou genetické změny u jedince způsobené mutagenezí probíhající v rodičovských gametách během oogeneze a spermatogeneze. Pojem „de novo“ by zde neměl být zaměňován s pojmem „nová mutace“. Přestože DNM v kontextu trojice (otec, matka a dítě) představují nové mutace, může se jednat o běžné, vzácné nebo nové varianty v obecné populaci. Pro měření a vysvětlení míry výskytu konkrétní DNM je nutné nejprve posoudit dopad varianty na fenotyp, protože nové příznivé znaky se mohou vyvinout, když vznikající genetické mutace nabízejí specifickou výhodu pro přežití (Front Line Genomics, 2017).
U lidí s genetickými nemendelovskými chorobami, které se vyskytují sporadicky, jsou DNM obvykle nové, spolehlivější a škodlivější než dědičné varianty, protože nepodléhají silnému přírodnímu výběru (Crow, 2000; Front Line Genomics, 2017). Identifikace genetické příčiny poruchy vyvolané DNM u jedince proto může být z klinického hlediska náročná, protože za jedním fenotypem může být pleiotropie a genetická heterogenita (Eyre-Walker a Keightley, 2007). V posledním desetiletí bylo proto vynaloženo značné úsilí na sekvenování exomů od jedinců s onemocněními nejasné genetické etiologie pro účely klinické diagnostiky. I po detekci kandidátních de novo variant však stále není k dispozici dostatek informací o běžných a vzácných variantách, což brání jasnému závěru o patogenitě identifikované de novo varianty a její roli v onemocnění (Acuna-Hidalgo et al., 2016). Toto omezení lze vysvětlit tím, že varianty de novo jsou obvykle heterozygotní a mohou být buď extrémně vzácné, nebo běžné. V případě velmi vzácných variant de novo může být obtížné prokázat patogenitu varianty, protože neexistuje více pacientů se stejným fenotypem a variantou de novo. V případech běžných de novo variant nemusí být známy faktory, které určují projevy patogenity varianty, zejména pokud někteří jedinci v běžné populaci mají danou variantu, ale nemají genetické onemocnění. Bez ohledu na míru výskytu variant de novo však lze oba typy variant škálovat na základě relativní zdatnosti a přirozeného výběru.
Adaptibilita závisí na mnoha faktorech, proto k posouzení, zda je DNM patogenní nebo adaptivní, a k pochopení, proč se v populaci vyskytuje s určitou frekvencí, je nutné zkoumat variantu za vhodných podmínek. Ty zahrnují prostředí, věk rodičů, genomický kontext, epigenetiku a další faktory, protože všechny ovlivňují hodnotu střední relativní zdatnosti, která monotónně roste, zatímco síla selekce klesá (Peck a Waxman, 2018).
Hlavním cílem této studie bylo objasnit míru výskytu DNM a zjistit, jak jsou tyto mutace distribuovány v exomech obecné litevské populace. Zkoumali jsme také, zda je četnost těchto mutací ovlivněna složením nebo strukturálními parametry sekvencí, v nichž se vyskytují, a dalšími faktory, které by mohly ovlivňovat mechanismy, jež stojí za vznikem těchto DNM. Nakonec jsme se snažili zjistit, zda DNM vznikly v důsledku intenzivního tlaku přírodního výběru na funkční oblasti. Ačkoli rozšíření a intenzita DNMs byly předmětem mnoha studií, v litevské populaci dosud zkoumány nebyly.
Materiál a metody
V této studii jsme analyzovali vzorky z litevské populace získané v rámci projektu LITGEN (LITGEN, 2011). Soubor dat se skládal ze 49 trojic s celkem 144 různými jedinci. Genomová DNA byla extrahována z venózní krve buď metodou fenol-chloroformové extrakce, nebo pomocí automatizované platformy pro extrakci DNA TECAN Freedom EVO® (Tecan Schweiz AG, Švýcarsko) založené na metodě paramagnetických částic. Exomy byly sekvenovány na sekvenačním systému SOLiD 5500 (75 bp čtení). Sekvenační data byla zpracována a připravena pomocí softwaru Lifescope. Exomy byly mapovány podle referenčního lidského genomu build 19. Průměrná hloubka čtení při sekvenování byla 38,5. Soubory ve formátu BAM matky, otce a dítěte vytvořené pomocí softwaru Lifescope byly pro každou trojici spojeny pomocí softwaru SAMtools.
Mutace de novo byly identifikovány pomocí dvou softwarových programů: VarScan (Koboldt et al., 2012) a VarSeqTM. Potenciální varianta byla považována za DNM, pokud byla identifikována u potomků, ale nebyla přítomna u žádného z rodičů na stejné pozici. Celkem bylo pomocí VarScan zjištěno 1 752 a pomocí VarSeqTM 4 756 DNM. K vyřazení falešně pozitivních volání de novo, kdy nebylo známo, zda byli všichni jedinci v trojici identifikováni správně, byly použity následující konzervativní filtry na zjištěné parametry kvality DNM: (1) kvalita genotypu jedince ≥50; (2) počet čtení na každém místě >20. K aplikaci těchto filtrů na data generovaná programem VarScan byl použit software SnpSift. Data vygenerovaná softwarem VarSeqTM byla filtrována výběrem stejných parametrů filtrování v segmentu Trio Workflow. Dále byly za účelem vyřazení zbývajících variant, které byly somatické (přítomné pouze ve zlomku sekvenovaných krvinek) s nízkým alelovým saldem nebo artefakty sekvenování, DNM filtrovány nastavením prahové hodnoty pro pozorovaný podíl čtení u jedinců s alternativní alelou (alelové saldo) pro trio (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). Kromě toho byly všechny možné identifikované a vyfiltrované jednonukleotidové varianty de novo ručně přezkoumány pomocí programu Integrative Genomics Viewer (Robinson et al., 2011). Vzhledem k velkému počtu identifikovaných DNM bylo pro ověření variant pomocí Sangerova sekvenování náhodně vybráno 51 jednonukleotidových variant de novo. Sangerovo sekvenování bylo provedeno pomocí genetického analyzátoru ABI PRISM 3130xl. Všechny filtrované a ručně revidované DNM identifikované pomocí VarScan (N = 95) a pomocí VarSeqTM (N = 84) byly anotovány pomocí programu ANNOVAR (Butkiewicz a Bush, 2016; Wang et al., 2010). Pro analýzu interakcí proteinů byl použit software STRING (Szklarczyk et al., 2017). Stejně jako v případě mapování exomu byly anotace provedeny pomocí referenčního lidského genomu hg19.
Pravděpodobnost, že volající pozice byla DNM v trojici, byla vypočtena nezávisle pro každou trojici. Jak bylo popsáno v předchozí referenci (Besenbacher a kol, 2015), byla míra de novo na pozici za generaci (PPPG) vypočtena takto:
kde f je počet trií a N je počet volaných míst, která lze potenciálně identifikovat jako de novo místa pro každou trojici zvlášť, bez ohledu na hloubku sekvenování. Tento počet se liší v závislosti na triu. ni je počet identifikovaných DNM pro trio i. Pravděpodobnost Pji (de novos ingle nukleotid) pro volané jediné nukleotidové místo j a rodinu i, které bude mutované, byla vypočtena následovně:
Pravděpodobnost Pji (de novo indel)pro volané indelové místo j a rodinu i, že bude mutováno, byla vypočtena takto:
kde C, M a F znamenají potomka, matku, resp. otce, a Hetero, HomR a HomA označují heterozygotní, homozygotní pro referenční, resp. homozygotní pro alternativní alelu. Pravděpodobnost Pij (de novo) byla vypočtena s ohledem na pokrytí sekvenováním. Intervaly spolehlivosti pro odhady míry byly vypočteny jako pro binomické podíly. Pro odhad míry DNM a pro další výpočty jsme použili balíček R (verze 3.4.3) (R Core Team, 2013).
Pro ověření hypotézy, že rozdíly v míře DNM v různých oblastech genomu lze vysvětlit vnitřními vlastnostmi samotné genomové oblasti a stářím rodičů, byla provedena lineární regresní analýza, pro kterou byla provedena „sekundární“ anotace každé DNM s využitím dat z projektů ENCODE (ENCODE Project Consortium, 2012) a LITGEN (LITGEN, 2011). Nejprve byly podle předchozí studie (Besenbacher et al., 2015) za účelem shromáždění záznamů týkajících se genomické krajiny identifikovaných DNM vybrány lymfoblastoidní buněčné linie (LCL a GM12878) (ENCODE Project Consortium, 2012). Byly shromážděny údaje o:
(1) míře exprese (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) v různých tkáních. Podle exprese byly oblasti s DNM rozděleny na pozice se specifickou a nespecifickou expresí;
(2) měření míst hypersenzitivity DNase1 (DHS). Status DHS byl přiřazen 0, pokud byl mimo vrchol DHS, a 1, pokud byl uvnitř;
(3) měření kontextu CpG ostrovů. Pokud se DNM nacházel uvnitř CpG ostrovů, byl mu přiřazen status pozice 1; pokud mimo – 0;
(4) tři histonové značky (H3K27ac, H3K4me1 a H3K4me3) z projektu ENCODE. Pokud se DNM nacházel v pozici označené histonem, byla mu přiřazena hodnota 1, pokud ne – 0;
(5) hodnoty zachování GERPP++ byly shromážděny pomocí anotačního nástroje ANNOVAR. Podle hodnot konzervace byly pozice s DNM rozděleny na konzervativní (GERP++ skóre >12) a nekonzervativní (GERP++ skóre <12) (Davydov et al., 2010; ENCODE Project Consortium, 2012). Na základě dotazníkových záznamů z projektu LITGEN byly shromážděny údaje o věku rodičů. Po shromáždění parametrů pro každou trojici byl vypočítán počet pozic s jednotlivými parametry. Poté byla provedena korelační analýza a následně lineární regresní modelování míry DNM a parametrů.
Výsledky
Po analýze DNM byl u dvou trojic (č. 4 a 21) identifikován mimořádně vysoký počet DNM: 113 a 123 (pomocí VarScan a VarSeqTM), resp. 16 (VarScan). Tato zjištění nás přiměla k provedení testu biologické paternity, který byl u trojice č. 1 zamítnut. 4 a potvrzeno u trojice č. 21. Údaje pro trojici č. 4 byla ze studie vyloučena. V konečném souboru 48 trojic bylo pomocí softwaru VarScan identifikováno 95 DNM u 34 trojic a pomocí softwaru VarSeqTM bylo identifikováno 84 DNM u 31 trojic (obrázek 1). Pomocí softwaru VarScan nebyly zjištěny žádné DNM u 18 a pomocí softwaru VarSeqTM u 15 trojic. Ze všech DNM identifikovaných oběma softwary se shodovalo pouze 5,37 % DNM (tři DNM v genech MEIS2, PGK1 a MT1B). Každá osoba měla v průměru 1,9 (software VarScan) a 1,7 (VarSeqTM) DNM.
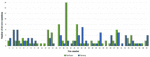
Obrázek 1. Srovnání jednonukleotidových variant de novo identifikovaných softwarem VarScan (modře) a VarSeqTM (zeleně).
Analýza 95 DNM, které byly identifikovány softwarem VarScan, ukázala, že 20 DNM bylo exonických, včetně dvou stop-gain DNM, sedmi synonymních DNM a 11 nesynonymních DNM. Osmdesát nových mutací identifikovaných pomocí softwaru VarSeqTM bylo exonických, včetně 1 DNM se stop-gain a 78 nesynonymních DNM (obrázek 2). Většina DNM identifikovaných systémem VarScan se nacházela na chromozomech 1, 2, 4 a 5, zatímco systém VarSeqTM identifikoval DNM převážně na chromozomech 2, 6, 7 a 11. Počet identifikovaných DNM nekoreloval s hustotou genů v chromozomech (R = 0,09, p-hodnota = 0,65 pro VarScan a R = 6,73, p-hodnota = 0,51 pro VarSeqTM) ani s velikostí chromozomu (obrázek 3). Podle obou softwarových programů byly poměry přechodů a transverzí velmi podobné: 1,44 a 1,47 (obrázek 4). Byly však zjištěny rozdíly ve struktuře přechodů. Konkrétně mezi DNM identifikovanými programem VarScan bylo více změn G/T a A/C, zatímco mezi DNM identifikovanými programem VarSeqTM bylo více změn A/T a G/C.
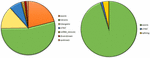
Obr. 2. Složení mutací de novo (DNM) vytvořených pomocí VarScan (vlevo) a pomocí VarSeqTM (vpravo).
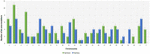
Obr. 3. Složení mutací de novo (DNM) vytvořených pomocí VarSeqTM (vpravo). Rozdělení počtu de novo variant podle chromozomů podle dat generovaných pomocí VarScan a VarSeqTM. Zelené sloupce představují DNM identifikované softwarem VarScan, modré – softwarem VarSeqTM. Chybové úsečky představují směrodatnou chybu průměrných DNM pro každý chromozom.
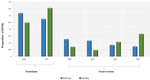
OBRÁZEK 4.
. Molekulární události, které jsou základem přechodů, se vyskytují častěji než události vedoucí k transverzím, což má za následek ∼1,5krát vyšší míru přechodů oproti transverzím v celém exomu. Události přechodu a transverze identifikované pomocí softwaru VarScan (zeleně) a VarSeqTM (modře). Chybové úsečky představují směrodatnou chybu průměrných DNM.
Vypočítané míry de novo jednonukleotidových mutací byly 2,4 × 10-8 PPPG (95% interval spolehlivosti : 1,96 × 10-8-2,99 × 10-8) podle softwaru VarSeqTM a 2,5 × 10-8 PPPG.74 × 10-8na nukleotid na generaci (95% CI: 2,24 × 10-8-3,35 × 10-8) podle VarScan.
Tři de novo indely ve třech trojicích byly identifikovány algoritmem VarScan na chromozómech 6 a 11. Na chromozómech 6 a 11 byly identifikovány tři de novo indely ve třech trojicích. Vypočtená míra de novo indelů v genomu byla 1,77 × 10-8 (95% CI: 6,03 × 10-9-5,2 × 10-8) PPPG. Pozoruhodné je, že všechny de novo indely byly „reverzibilní“, tj. rodiče měli nové varianty v genomu a jejich děti měly de novo varianty založené na referenčním genomu se střední hodnotou hloubky sekvenování 37,5, resp. 50 genotypů kvality. Tyto tři DNM však nebyly vybrány pro validaci metodou Sangerova sekvenování, a tak přesto zůstává pravděpodobnost nadhodnocení de novo indelů. De novo indely byly v kontextu jednotlivých nukleotidů C/T a A/G.
Lineární regresní modelování ukázalo, že místa hypersenzitivity DNAse 1, kontext ostrovů CpG, hodnoty zachování GERPP++ a úrovně exprese vysvětlují ∼68-93 % míry DNM (tabulka 1). Epigenetické markery ani věk otce s mírou DNM významně nekorelovaly. Modely byly stanoveny pouze z údajů získaných ze systému VarScan, protože mezi údaji ze systému VarSeqTM a vlastními charakteristikami samotné genomové oblasti nebyla zjištěna žádná korelace.

TABULKA 1. V tabulce 1 je uvedeno, jaká je míra DNM. Lineární regrese vlivu míst hypersenzitivity DNAaseI, kontextu ostrovů CpG, hodnot zachování GERPP++ a úrovně exprese na míru DNMs.
Funkční predikce DNMs
S cílem posoudit, které missense mutace byly škodlivé a měnily funkci postiženého proteinu podle typu, bylo analyzováno predikované kategoriální skóre pro poškození vyvolané DNMs. V úvahu bylo bráno následujících 10 hodnot: polyfen HDIV a HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, kódování Fathmm-MKL a GERP++. Na základě předpovězených skóre byly vybrány čtyři DNM, které byly programem VarScan identifikovány jako ty, které mají šest nebo více poškozujících nebo pravděpodobně poškozujících předpovědí. Tyto stop-gain DNM byly v genech MEIS2 a ULK4, zatímco nesynonymní DNM byly v genech MT1B a PGK1. Proteiny kódované těmito geny jsou důležité pro růst neuronů, endocytózu a ochranu před negativními účinky těžkých kovů. Tyto proteiny se podílejí na uvolňování inhibitoru nádorových cév angiostatinu a na různých signálních drahách. Mezi proteiny kódovanými těmito geny nebyly zjištěny žádné souvislosti (obrázek 5).
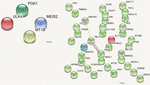
OBRÁZEK 5. Interakce protein-protein (Szklarczyk et al., 2017) u genů nesoucích DNM. DNM identifikované pomocí VarScan v genech kódujících proteiny jsou vlevo, DNM identifikované pomocí VarSeqTM – vpravo. Barevné čáry označují spojení mezi proteiny.
De novo mutace identifikované pomocí VarSeqTM byly podrobněji analyzovány, pokud byly alespoň polovinou predikčních nástrojů předpovězeny jako škodlivé nebo pravděpodobně škodlivé. Bylo zjištěno 35 bodových mutací (viz ??) v genech kódujících proteiny důležité pro remodelaci chromatinu, regulaci cytoskeletu, růst a životaschopnost buněk, cytoplazmatické signální dráhy a iniciaci neuronálních reakcí spouštějících vnímání pachu.
Mezi proteiny kódovanými geny postiženými DNM byly určitým způsobem propojeny pouze geny CLPTM1, ZNF547 a DMXL1 (obrázek 5).
Diskuse
V této studii jsme provedli komplexní analýzu distribuce DNM v různých oblastech exomu u litevské populace. Pomocí sekvenační technologie SOLiD 5500 bylo pomocí algoritmů VarScan a VarSeqTM zjištěno celkem 95 DNM ve 34 trojicích a 84 DNM ve 31 trojicích. Nejprve bychom chtěli poznamenat, že jsme pro volání DNMs zvolili VarScan, protože podle (Warden et al., 2014) je výsledkem tohoto algoritmu seznam variant, který má vysokou shodu (>97 %) s kvalitními variantami volanými pomocí GATK UnifiedGenotyper a HaplotypeCaller. Software VarSeqTM byl vybrán, protože se jedná o široce používaný nástroj pro analýzu variant jak ve výzkumu, tak v klinické analýze. Přestože jsou oba algoritmy navrženy tak, aby v exomu potomka hledaly DNM, které nebyly přítomny u žádného z rodičů, shoda mezi oběma softwary pro analýzu DNM byla pouze 5,37 %. Algoritmus VarScan měl vyšší citlivost (5,42 %) pro detekci DNM před filtrací než algoritmus VarSeqTM (1,77 %), měli jsme tedy podezření, že žádný nástroj neuspěl při vyvolávání mutací kvůli vysoké citlivosti, která byla vždy doprovázena nízkou specificitou. Proto navrhujeme, že výrazného zlepšení výsledků by mohlo být dosaženo kombinací výstupů různých nástrojů (Sandmann et al., 2017).
Na základě vytvořených dat byla odhadovaná míra jednonukleotidových DNM mezi 2,4 × 10-8 a 2,74 × 10-8 a míra de novo indelů byla 1,77 × 10-8 PPPG v závislosti na použitém algoritmu. Námi vypočtená míra DNM byla vyšší než míra uvedená v předchozích studiích (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015), ve kterých se pohybovala mezi 1,2 × 10-8 a 1,5 × 10-8 PPPG. Vyšší míra DNM v naší studii byla přiměřená, protože naše studie byla založena na datech z exomu. Navíc exomy vykazují výrazně vyšší (o 30 %) míru mutací než celé genomy, protože složení párů bází v celém genomu je jiné než v exomech. Zejména exomy mají průměrný obsah GC přibližně 50 %, zatímco u celého genomu je to přibližně 40 % (Neale et al., 2012). Metylizované CpG představují u člověka vysoce mutabilní sekvence v důsledku spontánní deaminace cytosinových bází (Neale et al., 2012). Podle studií srovnávací genomiky se předpokládá, že zvýšená míra mutací v oblastech bohatých na CpG se vyvinula přibližně v době radiace savců (Francioli et al., 2015). Během divergence druhů prošly exonické oblasti bohaté na CpG zvýšenou mírou mutací ve srovnání s oblastmi u nekódující DNA a změnily se v nekódující oblasti. Proto se pak vliv obsahu CpG v průběhu času snižuje, průměrná míra mutací klesá, až dosáhne úrovně přítomné v okolní nekódující DNA (Subramanian a Kumar, 2003). Zatímco však sekvence v neutrálně se vyvíjejících oblastech genomu měly dostatek času, aby se vyrovnaly s ohledem na dinukleotidové kontexty, čistící selekce udržovala hypermutabilní CpGs ve funkčních oblastech (Subramanian a Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Protože jsme tedy zjistili vyšší míru DNM, než uvádějí jiné studie, spekulovali jsme, že by to mohlo být alespoň částečně způsobeno lokálním sekvenčním kontextem a/nebo možným tlakem přirozené selekce na exom. V souladu s tím byl použit lineární regresní model a zjistili jsme, že hypersenzitivita DNAse 1, kontext ostrovů CpG, hodnoty zachování GERPP++ a úroveň exprese vysvětlují ∼68-93 % míry DNM. Tato zjištění naznačila, že DNM v exomu vznikají nezávisle na zachování sekvencí DNA. Míra DNM však byla vyšší u genů, jejichž produkty byly nespecifické, a v transkripčně aktivních promotorových oblastech.
Na rozdíl od výsledků jiných studií (Wong et al., 2016; Sandmann et al., 2017) jsme zjistili, že věk otce nekoreluje s mírou DNM. Tato zjištění lze vysvětlit tím, že soubor dat se skládal z trojic s podobným věkem rodičů a že byla analyzována pouze malá část (∼1,5 %) celého genomu. Na základě těchto parametrů měl každý člověk v průměru pouze 1,9 (VarScan) nebo 1,7 (VarSeqTM) DNM ve srovnání se 40-82 DNM v celém genomu (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al., 2016), zatímco počet de novo indelů v kódující sekvenci byl podobný jako v (Front Line Genomics, 2017).
Výsledky naší rozsáhlé funkční analýzy anotací ukázaly, že ze všech identifikovaných DNM byly 4 (VarScan) a 35 (VarSeqTM) variant pravděpodobně patogenní DNM. Rozdíl v počtu patogenních DNM lze vysvětlit tím, že v závislosti na algoritmu použitém pro identifikaci DNM se podíl DNM v kódujících sekvencích výrazně lišil. Například 21,05 % DNM identifikovaných softwarem VarScan bylo exonických, zatímco 95,24 % DNM identifikovaných softwarem VarSeqTM bylo exonických. Tyto patogenní DNM byly v genech kódujících proteiny, které jsou nezbytné pro modelování chromatinu, regulaci cytoskeletu, modulaci růstu a vitality buněk, funkci cytoplazmatických signálních drah a iniciaci neuronální odpovědi. Přestože jsou tyto DNM považovány za patogenní, všichni jedinci účastnící se průzkumu se označili za geneticky „zdravé“. Tento výsledek tedy naznačoval, že navzdory domnělé patogenitě DNM genomy, v nichž se DNM nacházely, tyto změny zřejmě tolerovaly, takže projevy onemocnění nebyly často výrazné. Podle Szamecze et al. (2014) platí, že čím častěji se DNM vyskytují v konzervovaných genetických pozicích, tím silnější je vliv přírodního výběru na genetické změny prostřednictvím kompenzačních mechanismů ochrany genomu. Škodlivé účinky variant lze zmírnit čtyřmi způsoby. Některé geny mohou tolerovat zkrácené varianty proteinů, protože jejich funkční účinky jsou maskovány neúplnou expresí, kompenzačními variantami nebo malým funkčním významem zkrácení (Bartha et al., 2015). Naproti tomu změny genů spojené s nesynonymními DNM jsou kompenzovány mechanismem akumulace užitečných mutací v celém genomu (Szamecz et al., 2014). To naznačuje, že v těchto případech nejsou patogenní mutace natolik škodlivé, aby snížily průměrnou zdatnost, a proto přetrvávají déle v mnoha generacích, přičemž jsou formovány přírodním výběrem.
Shrnuto, naše analýza distribuce DNMs a jejich genetického a epigenetického kontextu poskytla vhled do genetické variability litevského genomu. Na základě těchto zjištění mohou další studie u skupin pacientů s genetickými chorobami usnadnit naši schopnost odlišit určité patogenní DNM od tolerovaných DNM na pozadí a identifikovat spolehlivé příčinné DNM. Hlavní omezení této studie však spočívalo v tom, že jsme nezkoumali variabilitu v nekódujících a regulačních oblastech genů. Tyto informace by mohly přispět k objasnění možných mechanismů vzniku DNM, které stále zůstávají nedostatečně jasné.
Accesní kódy
Data o sekvencích byla uložena v Evropském nukleotidovém archivu (ENA) pod přístupovým číslem PRJEB25864 (ERP107829).
Etické prohlášení
Tato studie byla provedena v souladu s doporučením povolení, Vilnius Regional Ethics Committee for Biomedical Research. Protokol byl schválen Vilniuskou regionální etickou komisí pro biomedicínský výzkum. Všechny subjekty poskytly písemný informovaný souhlas v souladu s Helsinskou deklarací.
Příspěvky autorů
LP provedl analýzu dat a připravil rukopis. AJ vypočítal míru de novo mutací. Sekvenování trojic exomů provedli LA a IK. Hlavním řešitelem byl VK.
Financování
Tato studie byla podpořena Evropským sociálním fondem v rámci opatření Globální grant. Projekt LITGEN č. VP1-3.1-ŠMM-07-K-01-013.
Prohlášení o střetu zájmů
Autoři prohlašují, že výzkum byl prováděn bez jakýchkoli komerčních nebo finančních vztahů, které by mohly být chápány jako potenciální střet zájmů.
Doplňkové materiály
Doplňkové materiály k tomuto článku naleznete online na adrese: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). Nové poznatky o vzniku a úloze de novo mutací ve zdraví a nemoci. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N. a další (2015). Charakteristika heterozygotních zkracujících variant proteinů v lidském genomu. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Nová variabilita a míra de novo mutací v celopopulačních de novo sestavených dánských trojicích. Nat Commun. 6:5969. doi: 10.1038/ncomms6969
PubMed Abstract | CrossRef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., and Rodin, S. R. (2010). Klastry ostrovů CpG a proepigenetická selekce pro CpG v exonech kódujících proteiny HOX a dalších transkripčních faktorů. Proc. Natl. Acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., and Bush, W. S. (2016). In silico funkční anotace genomových variací. Curr. Proto se v roce 2016 konala konference genomů. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Původ, zákonitosti a důsledky lidských spontánních mutací. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract | CrossRef Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifikace vysoké části lidského genomu, která je pod selekčním omezením, pomocí programu GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | CrossRef Full Text | Google Scholar
ENCODE Project Consortium (2012). Integrovaná encyklopedie elementů DNA v lidském genomu. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., and Keightley, P. D. (2007). The distribution of fitness effects of new mutations [Rozložení účinků nových mutací na fitness]. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract | CrossRef Full Text | Google Scholar
Francioli, L. C., Polak, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genomové vzorce a vlastnosti de novo mutací u lidí. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | CrossRef Full Text | Google Scholar
Front Line Genomics (2017). Front Line Genomics Magazine Issue 14 – ASHG. Londýn: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Genetické vlivy na expresi genů napříč lidskými tkáněmi. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | CrossRef Full Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: odhalování somatických mutací a změn počtu kopií u nádorových onemocnění pomocí sekvenování exomu. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Míra de novo mutací a význam věku otce pro riziko onemocnění. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Jemné rozdíly v rychlosti rekombinace mezi pohlavími, populacemi a jedinci. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | CrossRef Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ‚t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Sekvenování transkriptomu a genomu odhaluje funkční variabilitu u lidí. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract | CrossRef Full Text | Google Scholar
LITGEN (2011). Dostupné na adrese: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders [Vzory a míra exonických de novo mutací u poruch autistického spektra]. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | CrossRef Full Text | Google Scholar
Peck, J. R., and Waxman, D. (2018). Co je to adaptace a jak by se měla měřit? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2013). Jazyk a prostředí pro statistické výpočty. Vídeň: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data (Hodnocení nástrojů pro volání variant pro neshodná data sekvenování nové generace). Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., and Sunyaev, S. (2008). Hypermutabilní nesynonymní místa jsou pod silnější negativní selekcí. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | CrossRef Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Neutrální substituce se v genomech primátů vyskytují rychleji v exonech než v nekódující DNA. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). Genomická krajina kompenzační evoluce Be. The genomic landscape of compensatory evolution [Genomická krajina kompenzační evoluce]. PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). Databáze STRING v roce 2017: kvalitativně kontrolované asociační sítě protein-protein, široce zpřístupněné. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: funkční anotace genetických variant z dat sekvenování nové generace. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., and Wu, X. (2014). Podrobné srovnání dvou populárních balíčků pro volání variant pro studie exomu a cílených exonů. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | CrossRef Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Nová pozorování vlivu věku matky na zárodečné de novo mutace. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | CrossRef Full Text | Google Scholar
.