Incidence a prevalence nemoci ledvinových kamenů v obecné populaci stále stoupá. Nefrolitiáza (NL) a nefrokalcinóza (NC), jejichž celoživotní prevalence dosahuje až 10 %, jsou proto zejména v západním světě velkou zdravotní zátěží (1). NL a NC jsou spojeny s významnou morbiditou a progresí do chronického onemocnění ledvin v důsledku recidivy, opakovaných chirurgických/endoskopických zákroků a souběžného zánětu. Na zjednodušené úrovni je tvorba ledvinových kamenů výsledkem nerovnováhy močových inhibitorů (např. citrátu, hořčíku, uromodulinu a pyrofosfátu) a promotorů (např. oxalátu, vápníku, fosfátu, urátu a cystinu) krystalizace, překročení přesycení s následnou agregací, nukleací a růstem kamenů na Randallově plaku (obr. 1). Tato nerovnováha může být způsobena změněnou enterální a/nebo renální manipulací buď s promotory, nebo s inhibitory, jako je enterální malsekrece oxalátu nebo renální malreabsorpce vápníku (obrázek 1).

Obrázek 1. Nerovnováha močových inhibitorů a promotorů krystalizace vedoucí k tvorbě ledvinových kamenů. Koncentrace močových inhibitorů a promotorů je ovlivňována a řízena střevními i ledvinovými transportéry (osa GI-ledviny). Tyto transportéry a výměníky, jako je SLC26A1, jsou zodpovědné za sekreci a absorpci. Pokud jde o oxalát, enterální hyperabsorpce, ale malsekrece a/nebo renální hypersekrece, ale malabsorpce vede k tomu, že hladina oxalátu v moči přesahuje přesycení, a tím podporuje krystalizaci a následnou tvorbu kamenů .
Předpokládá se, že základní etiologie NL je multifaktoriální s environmentální, významnou dietní, hormonální a genetickou složkou. Ve studiích dvojčat byla dědičnost ledvinových kamenů odhadnuta na 56 % (3) a až dvě třetiny osob tvořících hyperkalciurické kameny mají příbuzné s NL (4). Přestože ledvinové kameny obsahující vápník tvoří více než 80 % všech, genetický základ těchto kamenů zůstává z velké části neznámý (5). S výjimkou variant v CLDN14, TRPV5, SLC34A1, ALPL, CASR a UMOD dosud celogenomové asociační studie nepřinesly podstatné genetické faktory (6-8). Byly však identifikovány rizikové alely v rámci genů, u nichž byl rovněž zjištěn přenos onemocnění na mendelovském základě, jako jsou CASR, SLC34A1 a SLC2A9 (9, 10). K dnešnímu dni bylo identifikováno více než 30 jednotlivých genů s online Mendelian Inheritance in Man-defined fenotypem, které se v případě mutace podílejí na vzniku NL/NC (tabulka 1).

Tabulka 1. Geny, o nichž je známo, že způsobují monogenní formy NL/NC.
Způsoby dědičnosti u monogenních forem zahrnují autozomálně dominantní, autozomálně recesivní a X-vázaný přenos. Zajímavé je, že u několika z těchto genů byl zaznamenán jak recesivní, tak dominantní způsob dědičnosti: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 a SLC4A1. Zatímco většina syndromických a závažných vrozených poruch vykazuje recesivní způsob dědičnosti (Bartter, Lowe, Dent, FHHNC a distální renální tubulární acidóza se senzorineurální hluchotou), mírnější stavy jsou spojeny spíše s mutacemi v dominantních genech. Většina kódovaných proteinů představuje renální transportéry solutů (např. SLC34A1, SLC34A3 a SLC9A3R1), ale u pacientů s NL/NC byly nalezeny také defektní chloridové kanály (CLCN5), proteiny těsného spojení (např. CLDN16/CLDN19) a metabolické enzymy (např. AGXT, APRT a CYP24A1). Základní porucha se tedy většinou nachází v tubulárním systému samotné ledviny, a proto ji lze připsat jako tubulopatii. Naopak a priori extrarenální stavy, jako je primární hyperoxalurie (PH), kde dysfunkce jaterních enzymů (AGXT, GRHPR a HOGA1) způsobuje hromadění oxalátů se sekundárním postižením ledvin, jsou myslitelnými příčinami NL/NC. Ačkoli se předpokládá, že každý fenotyp onemocnění představuje relativně vzácnou jednotku, příčiny s jedním genem mohou díky své široké genetické heterogenitě představovat významný počet pacientů (42). Kromě genetické heterogenity existuje také alelická variabilita, kdy zkracující varianty vedou spíše ke ztrátě funkce a missense varianty (hypomorfy) mohou způsobovat spíše subtilní defekty, které lze klinicky přehlédnout, zejména u dospělých kamenů. Další nedávno doceněný fenomén se týká účinků dávkování genů u několika výše zmíněných genů pro ledvinové kameny. Například u genu SLC34A3, kódujícího jeden z hlavních transportérů fosfátů v proximálním tubulu (NaPiIIc), se ukázalo, že heterozygotní jedince již nelze považovat pouze za zdravé přenašeče, neboť se u nich kalcifikace ledvin a/nebo kostní projevy vyskytují podstatně častěji než u jedinců divokého typu; stále však v menší míře než u jedinců bialelických (homozygotních a složených heterozygotů) (43). Podobná pozorování byla zaznamenána u rodin s mutacemi v CYP24A1 (44). Podíl monogenních poruch na celkové prevalenci onemocnění ledvinovými kameny nebyl v minulosti komplexně studován. Zejména chybí genetické důkazy založené na rozsáhlém screeningu mnoha kauzálních genů u velkých kohort pacientů. Komplexní genetické testování bylo v minulosti příliš nákladné a neefektivní. Pro většinu jedinců s NL/NC proto nebyla mutační analýza příčinné genetické vady dostupná, přestože znalost molekulární příčiny NL/NC může mít důležité důsledky pro prognózu, profylaxi a/nebo léčbu. Ze studií klinického pozorování byly odvozeny pouze hrubé odhady: na základě rozsáhlého souboru dat z analýzy složení kamenů byl učiněn závěr, že monogenní příčiny nepřesahují 9,6 % u dětí a 1,6 % u dospělých (45). V posledním desetiletí se však tato situace začala měnit s nástupem technik vysokokapacitního sekvenování.
Vysokokapacitní analýza mutací u pacientů s NL/NC
Pro vyšetření pacientů s onemocněním ledvinových kamenů na přítomnost patogenních mutací ve známých genech onemocnění jsme vytvořili genový panel založený na mikrofluidní multiplex-PCR a konsekutivním sekvenování NextGen (Fluidigm™/NGS) (46, 47).
V „pilotní studii“ jsme postupně rekrutovali 268 geneticky nevyhraněných jedinců z typických klinik pro léčbu ledvinových kamenů; 102 dětských a 166 dospělých probandů. Díky tomu jsme identifikovali 50 delečních variant ve 14 z 30 analyzovaných genů, což vedlo k molekulární diagnóze u 15 % všech případů. V dětské podskupině jsme zjistili kauzální mutaci u 21 %, zatímco mezi dospělými byly deleční varianty přítomny u 11 % (obrázek 2A) (48). V kohortě dospělých byly nejčastěji nalezeny mutace v genu pro cystinurii SLC7A9 (obrázek 2B). Tyto výsledky se podařilo potvrdit ve dvou navazujících studiích. Za prvé, ve výhradně pediatrické kohortě 143 pacientů s NL/NC bylo 17 % případů vysvětleno mutacemi ve 14 různých genech (49). Za druhé, v kohortě 51 rodin s věkem manifestace NL/NC před 25 lety byla pomocí cílené WES odhalena genetická příčina téměř ve 30 % (50). Není překvapením, že recesivní mutace byly častěji nalezeny u novorozenců a v případech vrozeného onemocnění, zatímco dominantní stavy se obvykle projevily později v životě. Tyto údaje naznačují, že genetická nemoc ledvinových kamenů je nedostatečně diagnostikovaným stavem, přestože molekulární diagnóza potenciálně ovlivní prognózu, profylaxi a/nebo léčbu. Omezením, které stojí za zmínku, je však potenciální výběrové zkreslení způsobené náborem ze specializovaných klinik pro léčbu ledvinových kamenů ve všech třech výše uvedených studiích.

Obrázek č. 2. Analýza mutací 30 známých genů pro monogenní nefrolitiázu (NL)/nefrokalcinózu (NC) u 268 pacientů s NL/NC. (A) Podíl monogenních příčin v podsouboru dětí a dospělých. (B) Počet monogenních příčin napříč geny (červená označuje dospělé; oranžová označuje dětské pacienty). Za zmínku stojí, že SLC7A9 byl nalezen nejčastěji mutovaný, zejména u mladých dospělých.
Identifikace nových genů lidských onemocnění pomocí přístupu kandidátních genů
Vysokokapacitní mutační analýza se používá také k vyhledávání patogenních variant v různých kandidátních genech. Jedním z nejzajímavějších nedávných zjištění byl objev lidských mutací v genu SLC26A1 (32). Od prvního popisu tvorby Ca-oxalátových (CaOx) ledvinových kamenů a NC u Slc26a1 (Sat1) -knockoutovaných myší Dawsonem et al. v roce 2010 je SLC26A1 v dobré víře kandidátem na NL gen (51). SLC26A1 kóduje aniontový výměník exprimovaný na bazolaterální membráně proximálních renálních tubulů, ilea a jejuna. Následně byly pomocí přístupu kandidátních genů identifikovány patogenní varianty u lidí s anamnézou časného nástupu CaOx-NL, konkrétně u dvou nepříbuzných jedinců s bialelickými missense variantami (32). Z funkčního hlediska byla patogenita identifikovaných variant prokázána in vitro, což vedlo k chybnému intracelulárnímu transportu a zhoršené transportní aktivitě (32). Defektní SLC26A1 proto představuje novou příčinu CaOx-NL a měl by být zvažován při testování jedinců na příčiny opakované tvorby CaOx-kamenů.
Nový klinický registr pacientů pro dědičnou ledvinovou kamennou nemoc
Většina epidemiologických údajů o zvyšující se prevalenci v západních zemích pochází z amerických databází. Ačkoli jsou naléhavě potřebné, centralizované evropské databáze nejsou v současné době k dispozici. Vzhledem k tomu, že výše uvedené genetické studie o prevalenci dědičné nemoci ledvinových kamenů byly provedeny s malými kohortami ze specializovaných center v Evropě i v USA, není převod na obecnou situaci v Evropě platný. Zatímco v USA představuje konsorcium Rare Kidney Stone Consortium platformu, která integruje a koordinuje aktivity v oblasti registru, základního a klinického výzkumu vzácných onemocnění, jako je cystinurie, PH, deficit APRT, Dentova a Loweho choroba, v Evropě ani v Německu dnes není realizován žádný srovnatelný sběr dat o pacientech s dědičnou chorobou ledvinových kamenů. Ve spolupráci se stávajícím evropským registrem PH, OxalEurope (prof. Bernd Hoppe, Univerzita v Bonnu), a díky financování Deutsche Forschungsgemeinschaft a Else Kröner-Fresenius Stiftung jsme nedávno na Univerzitě v Lipsku založili „Registr klinických pacientů pro dědičnou nemoc ledvinových kamenů“. Registr je celostátně podporován německými společnostmi pro nefrologii dospělých (DGfN) a dětskou nefrologii (GPN). Dále je zapsán v německém registru klinických studií (DRKS-ID: DRKS00012891). Jako základní součást náboru do studie je na výzkumné bázi nabízena vysokokapacitní analýza mutací známých a nových genů pro ledvinové kameny u pacientů bez stanovené molekulární diagnózy, ale s klinickým obrazem, který ukazuje na základní genetickou náchylnost: např. časný věk nástupu (<40 let), pozitivní rodinná anamnéza, orientační fenotypy jako NC, cystinurie nebo RTA a těžce recidivující NL (>3×) (tabulka 2). Zatímco pacienti s již stanovenou genetickou diagnózou jsou zpravidla zařazováni, případy se sekundárními příčinami NL/NC, jako jsou malignity, sarkoidóza a primární hyperparatyreóza, se do genetické analýzy nezařazují. K aktivnímu zařazení pacientů potřebuje klinické centrum obvykle souhlas místní Institutional Review Board; tento proces, při kterém nabízíme pomoc a asistenci poskytnutím příslušných šablon. Po etickém schválení lze formulář souhlasu a listy s klinickými údaji (např. klinický dotazník) stáhnout z našich webových stránek registru (http://www.mks-registry.net). Abychom zajistili důkladnou klinickou fenotypizaci, budeme žádat o podstatné informace o pacientech, jako je etnický původ, příbuzenství, rodinná anamnéza, věk vzniku, recidiva (definovaná jako každá domněle nová příhoda ledvinových kamenů), denní příjem tekutin, chirurgické zákroky a mimo jiné i extrarenální postižení. Dokumenty může vyplnit pacient s pomocí registrujícího lékaře. Kromě toho budou při zařazení požadovány biochemické parametry v séru, včetně kreatininu, eGFR, PTH, vitaminu D, elektrolytů, kyseliny močové a vyšetření moči (pH, vápník, fosfát, hořčík, kyselina močová, citrát, oxalát a cystin, nejlépe z 24hodinové moči, pokud se nejedná o moč bodovou), a také údaje o analýze složení kamenů. Vzhledem k tomu, že sběr 24hodinové moči a analýza složení kamenů se neprovádí rutinně ve všech institucích, zahrneme tyto parametry podle dostupnosti. Dvouleté klinické kontroly zařazených pacientů jsou žádoucí, ale nejsou povinné. Po registraci bude přijímajícím klinickým centrům poskytnuto osobní přihlašovací jméno pro zadávání údajů o pacientech prostřednictvím našich webových stránek registru (http://www.mks-registry.net). Případně nabízíme zadávání údajů elektronicky, pokud nám budou zaslány v papírové podobě. Zadané údaje budou uloženy na zabezpečeném serveru a zúčastněná klinická centra k nim budou mít přístup, aby si mohla prohlédnout vlastní údaje o pacientech. Další informace poskytují následující webové stránky:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
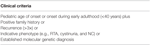
Tabulka 2. Kritéria pro zařazení do mutační analýzy v klinickém registru pacientů.
Shrnem lze říci, že onemocnění ledvinovými kameny je stále častěji se vyskytujícím onemocněním, které je klinicky heterogenní a málo známé, zejména jeho genetické příčiny. Jak naznačila řada nedávných studií, prevalence monogenních stavů je s největší pravděpodobností podhodnocena. Zavedením centralizovaného registru pacientů s dědičným onemocněním ledvinových kamenů přispějeme k alespoň částečnému překonání rozsáhlé mezery ve znalostech o genetice onemocnění ledvinových kamenů. V této souvislosti jsou klinické registry cenným zdrojem z několika důvodů: zaprvé, vymezení lepších asociací mezi fenotypem a genotypem bude mít zásadní význam pro přesnější stratifikaci pacientů v budoucích klinických výzkumných studiích. Za druhé, identifikace nových genů onemocnění s novými mechanismy onemocnění zmenší mezeru neznámé etiologie NL/NC; a za třetí, rozluštění nových molekulárních cílů pomůže připravit půdu pro vývoj léků prevence recidivy u těžce postižených rodin.
Etické prohlášení
Tato studie byla provedena v souladu s doporučeními „Ethikkommission an der Medizinischen Fakultät der Universität Leipzig“ s písemným informovaným souhlasem všech subjektů. Všechny subjekty poskytly písemný informovaný souhlas v souladu s Helsinskou deklarací. Protokol byl schválen „Ethikkommission an der Medizinischen Fakultät der Universität Leipzig.“
Příspěvek autora
JH vymyslel a napsal rukopis. BH, AS, LM a RS upravili rukopis a vytvořili infrastrukturu registru, který je čtenáři představen.
Prohlášení o střetu zájmů
Autoři prohlašují, že výzkum byl prováděn bez jakýchkoli komerčních nebo finančních vztahů, které by mohly být chápány jako potenciální střet zájmů.
Financování
Výzkum je financován z projektových grantů DFG (HA 6908/2-1) a EKFS (2016_A52) pro JH. Tato práce byla dále podpořena Spolkovým ministerstvem pro vzdělávání a výzkum (BMBF), Německo, FKZ: 01EO1501 pro JH.
1. Scales CD Jr, Smith AC, Hanley JM, Saigal CS. Prevalence ledvinových kamenů ve Spojených státech. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nefrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nefrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Genetika hyperkalciurické nefrolitiázy: onemocnění ledvinnými kameny. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Srov. např. Sayer JA. Pokrok v pochopení genetiky nefrolitiázy s obsahem vápníku. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576.
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 ovlivňuje koncentraci kyseliny močové s výrazným vlivem na pohlaví. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutace v genu SLC2A9 pro transportér glukózy 9 způsobují renální hypourikémii. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identification and characterization of a gene with base substitutions associated with the absortive hypercalciuria phenotype and low spinal bone density. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Chybné cílení peroxisomální L-alanin:glyoxylát aminotransferázy na mitochondrie u pacientů s primární hyperoxalurií závisí na aktivaci kryptické mitochondriální cílové sekvence bodovou mutací. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Lidská adeninfosforibosyltransferáza. Identifikace alelických mutací na úrovni nukleotidů jako příčina úplného nedostatku enzymu. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutace v ATP6N1B, kódující novou podjednotku 116-kD vakuolární protonové pumpy ledvin, způsobují recesivní distální renální tubulární acidózu se zachovaným sluchem. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutace v genu kódujícím podjednotku B1 H+-ATPázy způsobují renální tubulární acidózu se senzorineurální hluchotou. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Syndrom nedostatku karboanhydrázy II v belgické rodině je způsoben bodovou mutací na invariantním histidinovém zbytku (107 His→Tyr): kompletní struktura normálního lidského genu CA II. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. A common molecular basis for three inherited kidney stone diseases. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutace v genu pro chloridový kanál CLCNKB způsobují Bartterův syndrom typu III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutace v těsném spojovacím genu claudin 19 (CLDN19) jsou spojeny s renálním úbytkem hořčíku, selháním ledvin a závažným postižením očí. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Gen kódující hydroxypyruvátreduktázu (GRHPR) je mutován u pacientů s primární hyperoxalurií typu II. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. Mutace HNF4A R76W způsobuje kromě fenotypu β buněk také atypický dominantní Fanconiho syndrom. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identifikace 17 nezávislých mutací zodpovědných za nedostatek lidské hypoxantin-guaninfosforibosyltransferázy (HPRT). Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetická heterogenita Bartterova syndromu odhalená mutacemi v K+ kanálu, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnion, přechodný antenatální Bartterův syndrom a mutace MAGED2. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Těsně propojené flankovací markery pro Loweův okulocerebrorenální syndrom s aplikací na hodnocení nosičství. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartterův syndrom, hypokalemická alkalóza s hyperkalciurií, je způsoben mutacemi v kotransportéru Na-K-2Cl NKCC2. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutace v SLC26A1 způsobují nefrolitiázu. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molekulární identifikace renálního urátového aniontového výměníku, který reguluje hladiny urátů v krvi. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nefrolitiáza a osteoporóza spojená s hypofosfatemií způsobenou mutacemi v sodíko-fosfátovém kotransportéru typu 2a. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familiární distální renální tubulární acidóza je spojena s mutacemi v genu pro aniontový výměník červených krvinek (Band 3, AE1). J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Cystinurie jiného typu než I způsobená mutacemi v SLC7A9, kódující podjednotku (bo,+AT) rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical type I xanthinuria. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Hildebrandt F. Genetická onemocnění ledvin. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutace v SLC34A3/NPT2c jsou spojeny s ledvinovými kameny a nefrokalcinózou. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Klinické a biochemické fenotypy dospělých s monoalelickými a bialelickými mutacemi CYP24A1: důkaz účinku dávky genu. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Récents acquis diagnostiques et thérapeutiques. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nefronophthis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identifikace 99 nových mutací v celosvětové kohortě 1 056 pacientů s ciliopatií související s nefronoftózou. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Fourteen monogenic genes account for 15% of nefrolithiasis/nephrocalcinosis. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalence monogenních příčin u dětských pacientů s nefrolitiázou nebo nefrokalcinózou. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing often detects a monogenic cause in early onset nefrolithiasis and nefrocalcinosis. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolitiáza a hepatotoxicita jsou spojeny s aniontovým transportérem Sat1 u myší. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar
.