Alexei Drummond, Andrew Rambaut, Remco Bouckaert, e Walter Xie
1 Introduzione
Questo tutorial introduce il software BEAST per l’analisi evolutiva bayesiana attraverso un semplice tutorial. Il tutorial prevede la co-stima della filogenesi di un gene e dei tempi di divergenza associati in presenza di informazioni di calibrazione da prove fossili.
Avrete bisogno del seguente software a vostra disposizione:
- BEAST – questo pacchetto contiene il programma BEAST, BEAUti, TreeAnnotator e altri programmi di utilità. Questo tutorial è scritto per BEAST v2.2.x, che ha il supporto per le partizioni multiple. È disponibile per il download da http://www.beast2.org/.
- Tracer – questo programma è usato per esplorare l’output di BEAST (e altri programmi MCMC bayesiani). Riassume graficamente e quantitativamente le distribuzioni dei parametri continui e fornisce informazioni diagnostiche. Al momento di scrivere, la versione corrente è v1.6. È disponibile per il download da
http://tree.bio.ed.ac.uk/software/. - FigTree – questa è un’applicazione per visualizzare e stampare filogenesi molecolari, in particolare quelle ottenute usandoBEAST. Al momento della scrittura, la versione attuale è v1.4.2. È disponibile per il download da http://tree.bio.ed.ac.uk/software/.
Questo tutorial vi guiderà attraverso l’analisi di un allineamento di sequenze prelevate da dodici specie di primati (vedi Figura 1). L’obiettivo è quello di stimare la filogenesi, il tasso di evoluzione di ogni stirpe e le età delle divergenze non calibrate.
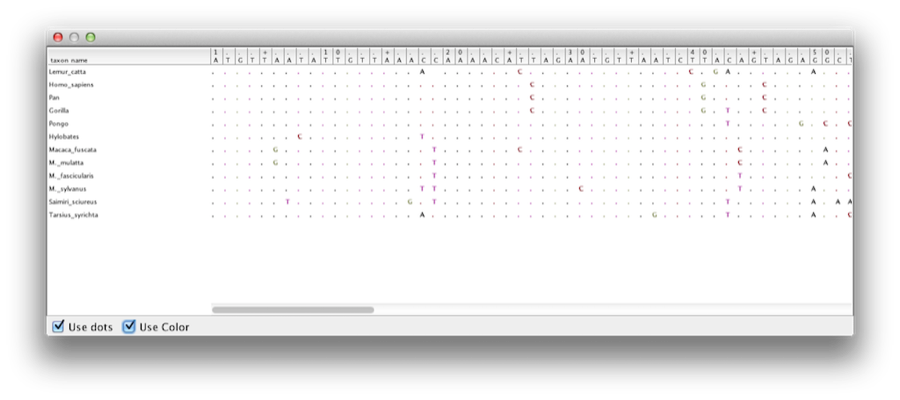
Figura 1: Parte dell’allineamento per i primati.
Il primo passo sarà convertire un file NEXUS con un blocco DATA o CHARACTERS in un file di input XML BEAST. Questo viene fatto utilizzando il programma BEAUti (che sta per Bayesian Evolutionary Analysis Utility). Questo è un programma facile da usare per impostare il modello evolutivo e le opzioni per l’analisi MCMC. Il secondo passo è quello di eseguire effettivamente BEAST utilizzando il file di input generato da BEAUTi, che contiene i dati, il modello e le impostazioni di analisi. Il passo finale è esplorare l’output di BEAST per diagnosticare i problemi e riassumere i risultati.
2 BEAUti
Il programma BEAUti è un programma facile da usare per impostare i parametri del modello per BEAST. Esegui BEAUti facendo doppio clic sulla sua icona. Una volta avviato, BEAUti avrà un aspetto simile a prescindere dal sistema su cui viene eseguito. Per questo tutorial, la versione Mac OS X è usata nelle figure, ma le versioni Linux e Windows avranno lo stesso layout e funzionalità.
2.1 Caricare il file NEXUS
Per caricare un allineamento in formato NEXUS, basta selezionare l’opzione Import Alignment… dal menu File, o trascinare il file al centro del pannello Partitions.
Il file di esempio chiamato primate-mtDNA.nex è disponibile nella directory examples/nexus/ per Mac e Linux e examples/nexus/ per Windows all’interno della directory dove è stato installato BEAST.Questo file contiene un allineamento di sequenze di 12 specie di primati.
Una finestra Add Partition (Figura 2) apparirà se il relativo pacchetto è installato. Se stai usando BEAST 2 “puro”, puoi andare al paragrafo successivo. Altrimenti, seleziona Add Alignment e clicca OK per continuare.
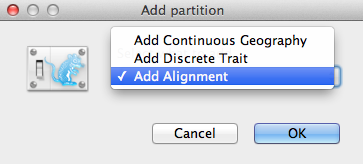
Figura 2: Finestra Add Partition (Appare solo se sono installati i pacchetti correlati).
Se c’è qualche sovrapposizione di codice nelle partizioni, apparirà la finestra del messaggio di avvertimento (Figura 3). Leggi e clicca OK per continuare.
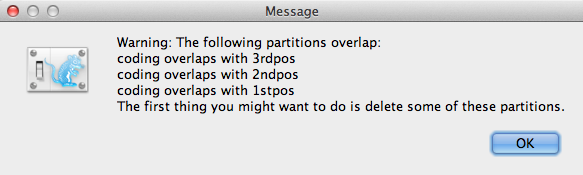
Figura 3: Finestra del messaggio di avvertimento (Appare solo se c’è qualche sovrapposizione di codifica nelle partizioni).
Una volta caricato, cinque partizioni di carattere sono visualizzate nel pannello principale (Figura 4). L’allineamento è diviso in una parte codificante della proteina e una parte non codificante, e la parte codificante è divisa nelle posizioni di codone 1, 2 e 3. Devi rimuovere la partizione ‘codificante’ prima di continuare al passo successivo perché si riferisce agli stessi nucleotidi delle partizioni ‘1stpos’, ‘2ndpos’ e ‘3rdpos’. Per rimuovere la partizione ‘codifica’ seleziona la riga e clicca sul pulsante ‘-‘ in fondo alla tabella. Potete visualizzare l’allineamento cliccando due volte sulla partizione.
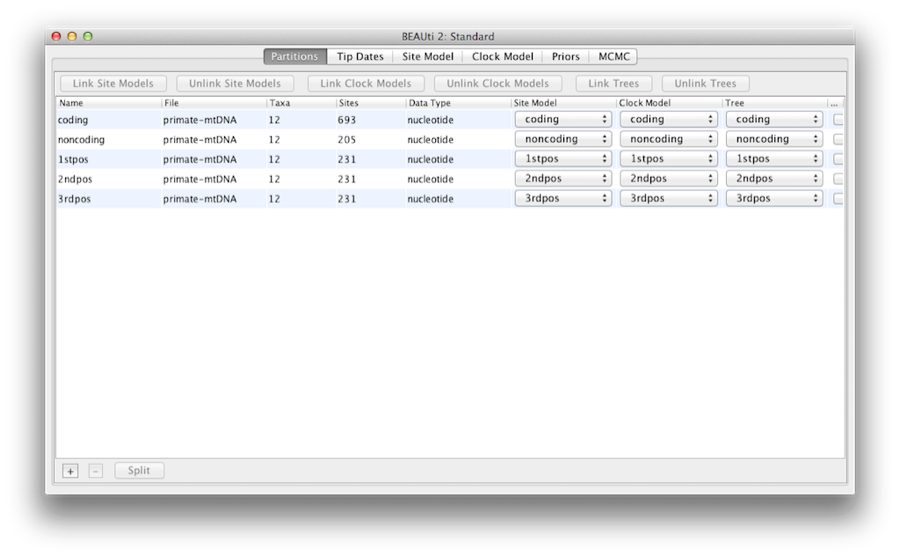
Figura 4: Una schermata della scheda dati in BEAUti. Questa e tutte le schermate seguenti sono state fatte su un computer Apple con Mac OS X e saranno leggermente diverse su altri sistemi operativi.
Link/Unlink partition models
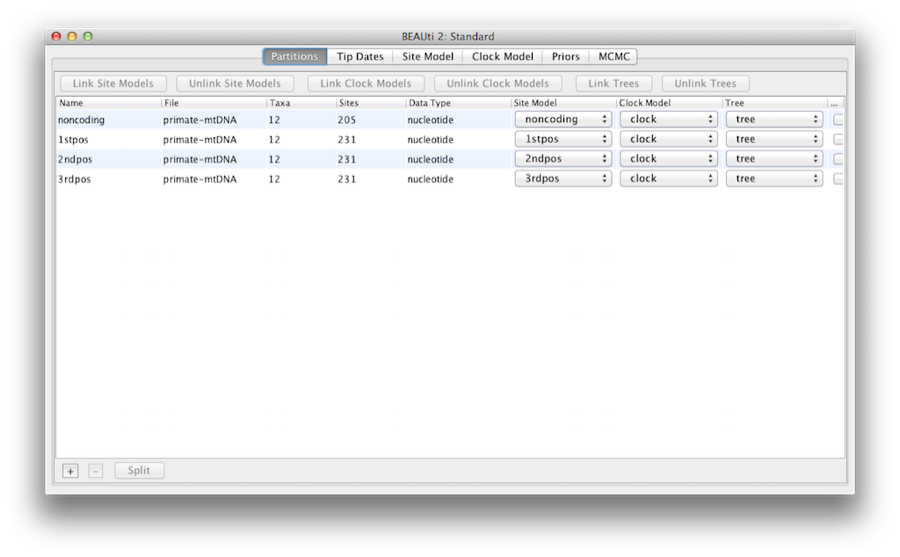
Figura 5: Una schermata della scheda Partitions in BEAUti dopo aver collegato e rinominato il modello di orologio e l’albero.
Siccome le sequenze sono collegate (cioè provengono tutte dal genoma mitocondriale, che si ritiene non subisca ricombinazione negli uccelli e nei mammiferi) condividono lo stesso antenato, quindi le partizioni dovrebbero condividere lo stesso albero temporale nel modello. Per semplicità, assumeremo anche che le partizioni condividano lo stesso tasso evolutivo per ogni ramo, e quindi lo stesso “modello di orologio”. Limiteremo la nostra modellizzazione dell’eterogeneità del tasso all’eterogeneità tra i siti all’interno di ogni partizione, e permetteremo anche alle partizioni di avere diversi tassi medi di evoluzione.
A questo punto dovremo collegare il modello di clock e l’albero. Nel pannello Partitions, selezionate tutte e quattro le partizioni nella tabella (o nessuna, per default tutte le partizioni sono interessate) e cliccate sul pulsante Link Trees e poi sul pulsante Link Clock Models (vedi Figura 5). Poi cliccate sul primo menu a discesa nella colonna Clock Model e rinominate il modello di clock condiviso in ‘clock’. Allo stesso modo rinominate l’albero condiviso in ‘tree’. Questo renderà le opzioni seguenti e i file di log generati più facili da leggere.
2.2 Impostare il modello di sostituzione
Il prossimo passo è impostare il modello di sostituzione. Quindi, selezionate la scheda Site Models nella parte superiore della finestra principale (saltiamo la scheda Tip Dates poiché tutti i taxa provengono da campioni contemporanei). Questo rivelerà le impostazioni del modello evolutivo per BEAST. Le opzioni disponibili dipendono dal fatto che i dati siano nucleotidi o amminoacidi, dati binari o dati generali. Le impostazioni che appariranno dopo aver caricato l’allineamento nucleotidico dei primati saranno i valori di default per i dati nucleotidici, quindi abbiamo bisogno di fare alcune modifiche.
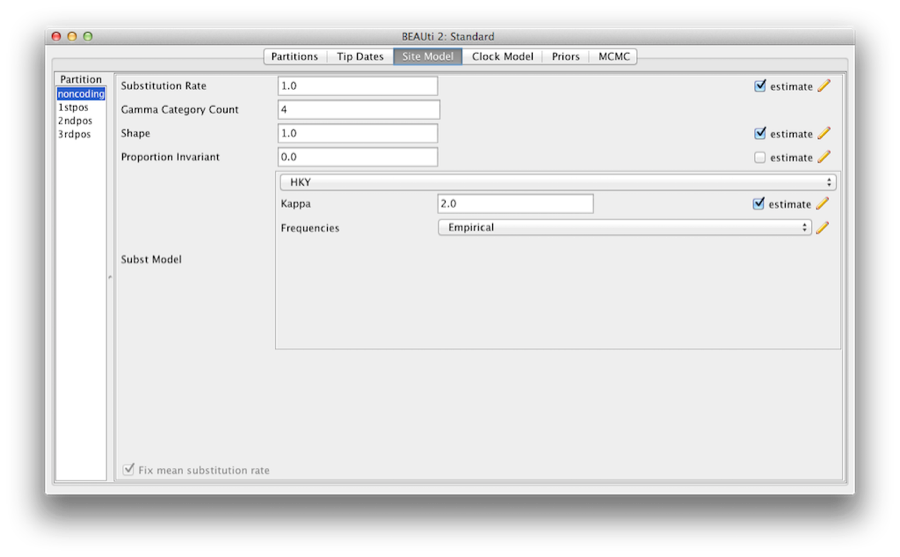
Figura 6: Una schermata della scheda del modello del sito in BEAUti.
La maggior parte dei modelli dovrebbe esservi familiare. Per prima cosa, imposta il conteggio della categoria Gamma a 4 e poi spunta la casella ‘stima’ per il parametro Shape. Questo permetterà di modellare la variazione di tasso tra i siti in ogni partizione. Nota che da 4 a 6 categorie funzionano sufficientemente bene per la maggior parte dei set di dati, mentre avere più categorie richiede più tempo per il calcolo con pochi benefici aggiunti. Lasciamo la voce Proportion Invariant impostata su zero.
Poi selezioniamo HKY dal menu a discesa Subst Model. Idealmente, dovrebbe essere selezionato un modello di sostituzione che si adatti meglio ai dati per ogni partizione, ma qui, per semplicità, usiamo HKY per tutte le partizioni. Inoltre, seleziona Empirical dal menu a discesa Frequencies. Questo fisserà le frequenze alle proporzioni osservate nei dati (per ogni partizione individualmente, una volta scollegati i modelli del sito). Questo approccio significa che possiamo ottenere un buon adattamento ai dati senza stimare esplicitamente questi parametri. Lo facciamo qui semplicemente per rendere i file di log un po’ più brevi e più leggibili nelle parti successive dell’esercizio.
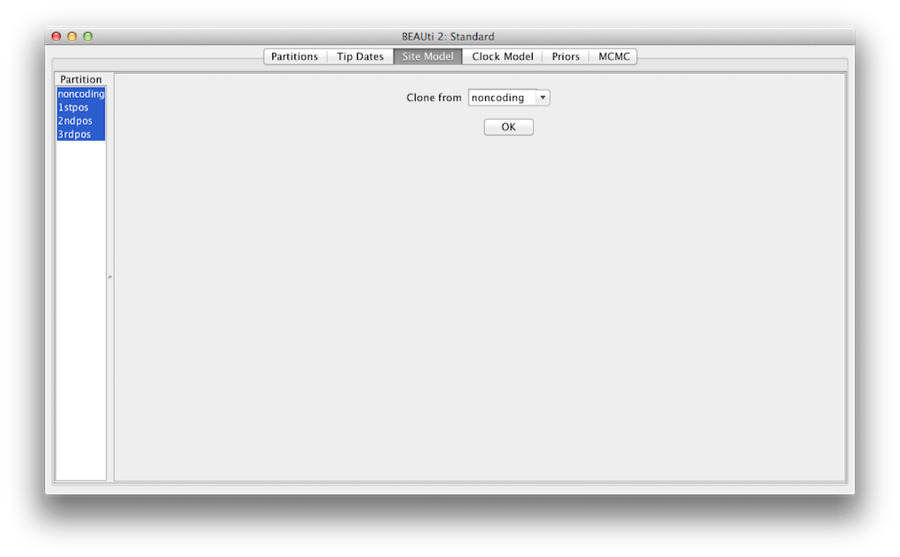
Figura 7: configurazione del clone da un modello di sito agli altri.
Finalmente spunta la casella ‘stima’ per il parametro Substitution rate e seleziona la casella Fix mean mutation rate. Questo permetterà alle singole partizioni di avere i loro tassi relativi stimati per i modelli di sito non collegati (Figura 6).
Infine, tenete il tasto ‘shift’ per selezionare tutti i modelli di sito sul lato sinistro, e cliccate su OK per clonare l’impostazione da noncoding in 1stpos, 2ndpos e 3rdpos (Figura 7). Passare attraverso ogni modello di sito, come si può vedere, le loro configurazioni sono le stesse ora.
2.3 Impostare il modello di orologio
Il prossimo passo è quello di selezionare la scheda Clock Models nella parte superiore della finestra principale. Qui è dove selezioniamo il modello di orologio molecolare. Per questo esercizio lasceremo la selezione al valore predefinito di un rigoroso orologio molecolare, perché questi dati sono molto simili all’orologio e non hanno bisogno di variazioni di tasso tra i rami per essere inclusi nel modello.
Per verificare l’attendibilità dell’orologio, è possibile (i) eseguire l’analisi con un modello di orologio rilassato e controllare quanta variazione tra i tassi è implicita nei dati (vedi coefficiente di variazione per saperne di più), o (ii) eseguire un confronto tra un modello di orologio rigoroso e uno rilassato utilizzando il pathsampling, o (iii) utilizzare un modello di orologio locale casuale che considera esplicitamente se ogni ramo dell’albero ha bisogno del proprio tasso di ramificazione.
2.4 Priori
La scheda Priori permette di specificare i priori per ogni parametro del modello. Le selezioni del modello fatte nelle schede modello del sito e modello dell’orologio, risultano nell’inclusione di vari parametri nel modello, e questi sono mostrati nella scheda dei priori (vedi Figura 8).
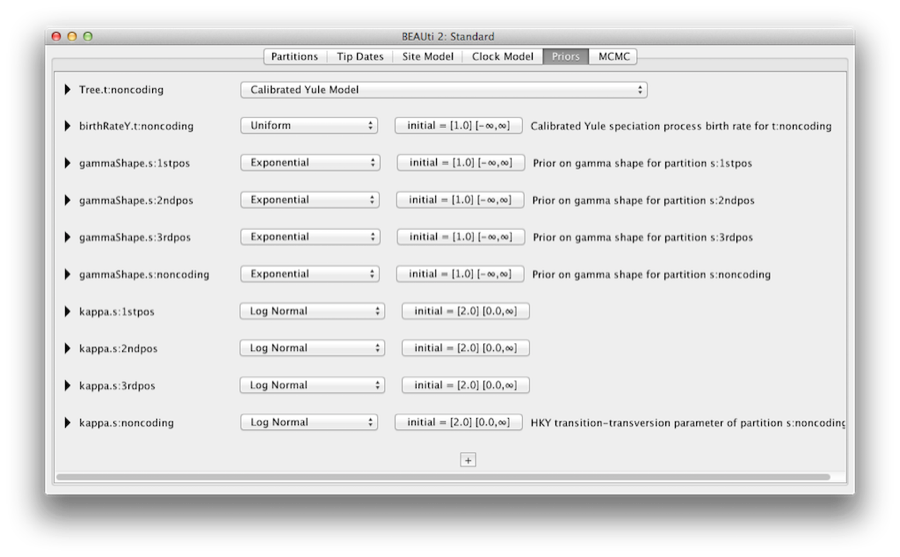
Figura 8: Una schermata della scheda Priori in BEAUti.
Qui specifichiamo anche che vogliamo utilizzare il modello di Yule calibrato come priore dell’albero. Il modello di Yule è un modello semplice di speciazione che è generalmente più appropriato quando si considerano sequenze di specie diverse. Selezionate il modello di Yule calibrato dal menu a discesa Priore dell’albero.
2.4.1 Definizione del nodo di calibrazione
Ora abbiamo bisogno di specificare una distribuzione priore sul nodo calibrato, basata sulla nostra conoscenza del fossile. Questo è noto come calibrazione del nostro albero. Per definire un ulteriore priore, premi il piccolo pulsante + sotto la lista dei priori. Se non è visibile nella tua vista, scorri il pannello fino in fondo per trovare il pulsante +. Vedrete una finestra di dialogo che vi permetterà di definire un sottoinsieme dei taxa nell’albero filogenetico. Una volta che avete creato un insieme di taxa, sarete in grado di aggiungere informazioni di calibrazione per il suo più recente commonancestor (MRCA) in seguito.
Nominate il taxa set compilando la voce dell’etichetta del taxon set. Chiamalo human-chimp, poiché conterrà i taxa di Homo sapiens e Pan. Nell’elenco sottostante vedrete i taxa disponibili. Selezionate a turno ciascuno dei due taxa e premete il pulsante freccia > >. (Figura 9).Cliccate su OK e l’insieme dei taxa appena definito verrà aggiunto alla lista dei precedenti.Poiché questo è un nodo calibrato da usare insieme al precedente Calibrated Yule, la monofilia deve essere applicata, quindi selezionate la casella di controllo segnata Monophyletic. Questo vincolerà la topologia dell’albero in modo che il raggruppamento uomo-scimpanzé sia mantenuto monofiletico nel corso dell’analisi MCMC.
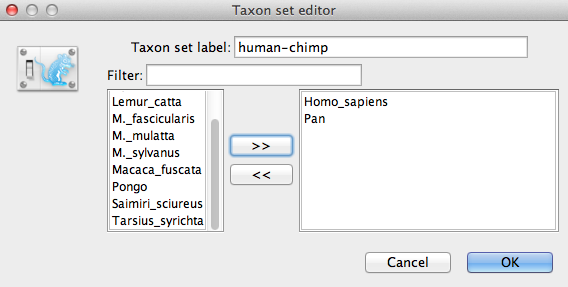
Figura 9: Editor del taxon set in BEAUti.
Per codificare le informazioni di calibrazione abbiamo bisogno di specificare una distribuzione per l’MRCA di human-chimp.Selezionare la distribuzione Log-normal dal menu a discesa a destra del nuovo human-chimp.prior. Cliccate sul triangolo nero e apparirà un grafico della funzione di densità di probabilità, insieme ai parametri per la distribuzione log-normale. Impostiamo M=1,78 e S=0,085 che specificherà una distribuzione centrata a circa 6 milioni di anni con una deviazione standard di circa 0,5 milioni di anni. Questo darà un intervallo centrale di probabilità del 95% che copre 5-7 Mya. Questo corrisponde all’incirca all’attuale stima consensuale della data dell’antenato comune più recente dell’uomo e dello scimpanzé (Figura 10).
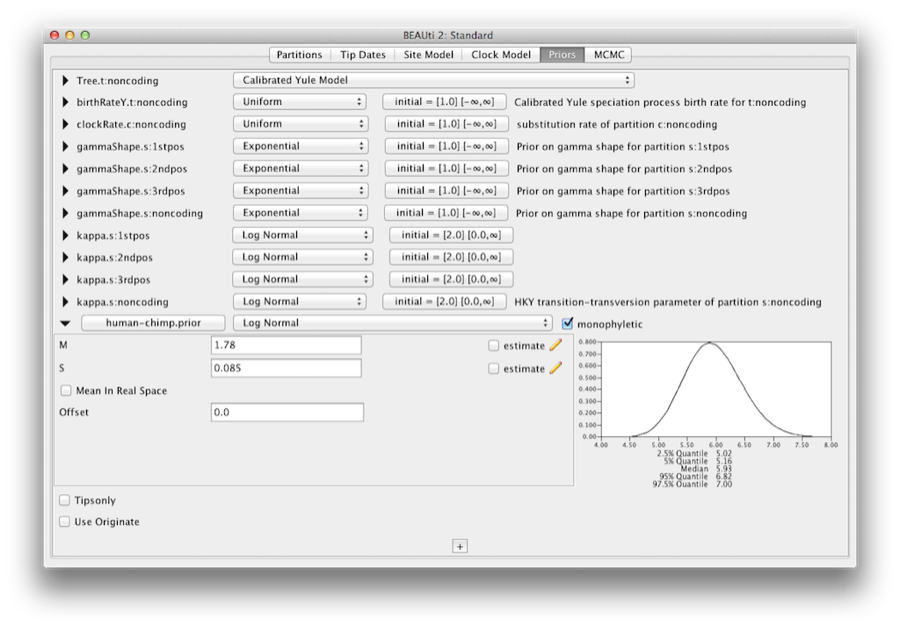
Figura 10: Uno screenshot delle opzioni di calibrazione delle priorità nel pannello Priors in BEAUti.
Dovremmo convincerci che i priori mostrati nel pannello dei priori riflettono realmente le informazioni a priori che abbiamo sui parametri del modello. Infine, specificheremo anche alcuni priori diffusi “disinformativi” ma appropriati sul tasso di clock molecolare complessivo (clockRate) e sul tasso di speciazione (birthRateY) del priore dell’albero di Yule. Per ciascuno di essi, selezionate Gamma dal menu a discesa e, utilizzando il pulsante freccia, espandete la vista per rivelare i parametri del priore Gamma. Sia per il tasso di clock che per il tasso di natalità di Yule impostate il parametro Alpha (forma) a 0,001 e il parametro Beta (scala) a 1000 (Figura 11).
Per default ognuno dei parametri di forma gamma ha una distribuzione priordine esponenziale con una media di 1. Questo implica (vedi Figura 3.7) che ci aspettiamo una certa variazione. Per default i parametri kappa per il modello HKY hanno una distribuzione a priori lognormale(1,1.25), che concorda ampiamente con l’evidenza empirica sull’intervallo di valori realistici per la transizione/transversione bias. Questi priori di default sono stati mantenuti in quanto adatti a questa particolare analisi.
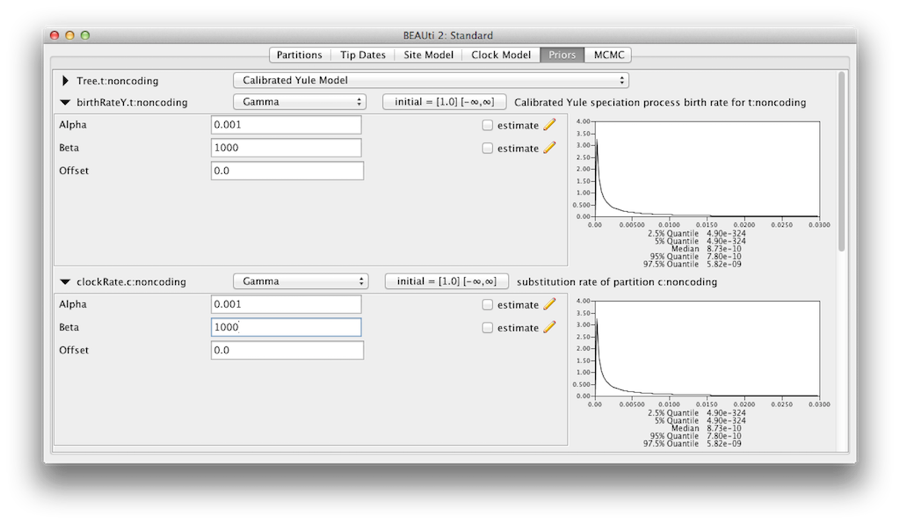
Figura 11: Priore gamma.
2.5 Impostare le opzioni MCMC
La prossima scheda, MCMC, fornisce impostazioni più generali per controllare la lunghezza della corsa MCMC e i nomi dei file.
Prima di tutto abbiamo la lunghezza della catena. Questo è il numero di passi che l’MCMC farà nella catena prima di finire. Quanto debba essere lungo dipende dalla dimensione del set di dati, dalla complessità del modello e dalla qualità della risposta richiesta. Il valore predefinito di 10.000.000 è del tutto arbitrario e dovrebbe essere regolato in base alle dimensioni del vostro set di dati. Per questo set di dati impostiamo la lunghezza della catena a 6.000.000, dato che questo funzionerà ragionevolmente veloce sulla maggior parte dei computer moderni (pochi minuti).
Il campo Store Every determina quanto spesso lo stato viene memorizzato su file. Memorizzare lo stato periodicamente è utile per situazioni in cui l’ambiente informatico non è molto affidabile e l’esecuzione di BEAST può essere interrotta. Avere una copia memorizzata dello stato recente ti permette di riprendere la catena invece di ricominciare dall’inizio, così non hai bisogno di ripetere il burn-in. Il campo Pre Burnin specifica il numero di campioni che non sono registrati all’inizio dell’analisi. Lasciamo i campi Store Every e Pre Burnin impostati ai loro valori di default. Sotto questi ci sono i dettagli dei file di log. Ognuno di essi può essere espanso cliccando sul triangolo nero.
Le prossime opzioni specificano quanto spesso i valori dei parametri nella catena di Markov dovrebbero essere visualizzati sullo schermo e registrati nel file di log.L’output dello schermo è semplicemente per monitorare il progresso dei programmi e può essere impostato su qualsiasi valore (anche se se impostato troppo piccolo, la quantità di informazioni visualizzate sullo schermo rallenterà effettivamente il programma). Per il file di log, il valore dovrebbe essere impostato in relazione alla lunghezza totale della catena. Campionare troppo spesso risulterà in file troppo grandi con poco beneficio extra in termini di accuratezza dell’analisi. Campionando troppo raramente, il file di log non registrerà informazioni sufficienti sulle distribuzioni dei parametri. Probabilmente vorrete puntare a memorizzare non più di 10.000 campioni, quindi questo dovrebbe essere non meno della lunghezza della catena / 10.000.
Per questo esercizio imposteremo la frequenza del trace log e del tree log a 1.000 e lo screen log a 10.000. Specificate anche Primates.log come nome del file del trace log e Primates.trees come nome del file del tree log.Assicurati che il nome del file del log dello schermo sia lasciato vuoto, o il log dello schermo non sarà scritto sullo schermo.
- Se stai usando il sistema operativo Windows allora ti suggeriamo di aggiungere il suffisso .txt a entrambi (quindi, Primates.log.txt e Primates.trees.txt) in modo che Windows li riconosca come file di testo.
2.6 Generazione del file BEAST XML
Siamo ora pronti per creare il file BEAST XML. Per farlo, seleziona l’opzione Save dal menu File. Controlla i priori di default, e salva il file con un nome appropriato (di solito finiamo il nome del file con .xml, cioè Primates.xml). Siamo ora pronti per eseguire il file attraverso BEAST.
3 Eseguire BEAST
Figura 12: Una schermata di BEAST.
Ora esegui BEAST e quando chiede un file di input, fornisci il tuo file XML appena creato come input. BEAST verrà poi eseguito finché non avrà finito di riportare le informazioni sullo schermo. I file dei risultati effettivi vengono salvati sul disco nella stessa posizione del tuo file di input. L’output sullo schermo sarà qualcosa di simile a questo:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Nota che ci sono alcune informazioni utili all’inizio riguardanti gli allineamenti e quali probabilità dell’albero sono usate. Inoltre, tutte le citazioni rilevanti per l’analisi sono menzionate all’inizio della corsa, che può essere facilmente copiata nei manoscritti che riportano l’analisi. Poi segue il resoconto della catena, che dà un feedback in tempo reale sul progresso della catena.
Alla fine, viene stampata un’analisi degli operatori, che elenca tutti gli operatori usati nell’analisi insieme a quanto spesso l’operatore è stato provato, accettato e rifiutato (vedi colonne #totale, #accetta e #rifiuta rispettivamente). Il tasso di accettazione è la proporzione di volte che un operatore viene accettato quando viene selezionato per fare una proposta. In generale, un tasso di accettazione che è alto, diciamo oltre 0,5 indica che le proposte sono conservative e non esplorano lo spazio dei parametri in modo efficiente. D’altra parte un basso tasso di accettazione indica che le proposte sono troppo aggressive e quasi sempre risultano in uno stato che viene rifiutato a causa del suo basso posteriore. Un tasso di accettazione di 0,234 è l’obiettivo (basato su prove molto limitate fornite da ) per molti (ma non tutti) gli operatori implementati in BEAST.
Alcuni operatori hanno un parametro di regolazione, per esempio il fattore di scala del parametro ascale. Se il tasso di accettazione finale non è vicino all’obiettivo, BEAST suggerisce un nuovo valore per il parametro di regolazione, che viene stampato nell’analisi dell’operatore. In questo caso, tutti i tassi di accettazione sono buoni per gli operatori che hanno parametri di regolazione. Gli operatori senza parametri di sintonizzazione includono gli operatori wideexchange e Wilson-Balding per questa analisi. Entrambi questi operatori tentano di cambiare la topologia dell’albero con grandi passi, ma poiché i dati supportano in modo schiacciante una singola topologia, queste proposte radicali sono quasi sempre respinte.
4 Analisi dei risultati
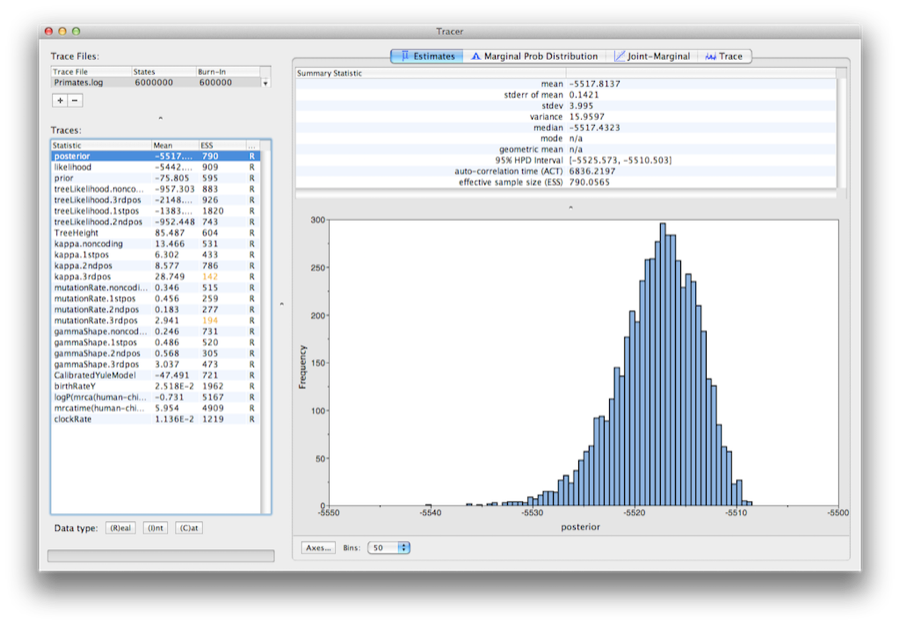
Figura 13: Una schermata di Tracer v1.6.
Lancia il programma chiamato Tracer per analizzare l’output di BEAST. Quando la finestra principale si è aperta, scegliete Import Trace File… dal menu File e selezionate il file cheBEAST ha creato chiamato Primates.log (Figura 13).
Ricordate che MCMC è un algoritmo stocastico quindi i numeri reali non saranno esattamente gli stessi di quelli rappresentati nella figura.
Sulla sinistra c’è una lista delle diverse quantità che BEAST ha registrato su file. Ci sono tracce per il posteriore (questo è il logaritmo naturale del prodotto della verosimiglianza dell’albero e la densità anteriore), e i parametri continui. Selezionando una traccia sulla sinistra si ottengono analisi per questa traccia sul lato destro, a seconda della scheda selezionata. Alla prima apertura, la traccia ‘posteriore’ è selezionata e varie statistiche di questa traccia sono mostrate sotto la scheda Estimates.In alto a destra della finestra c’è una tabella di statistiche calcolate per la traccia selezionata.
Selezionate il parametro clockRate nell’elenco a sinistra per guardare il tasso medio di evoluzione (medio su tutto l’albero e su tutti i siti). Tracer traccerà un istogramma (marginale posteriore) per la statistica selezionata e vi darà anche statistiche riassuntive come la media e la mediana. Il 95% HPD sta per highest posterior density interval e rappresenta l’intervallo più compatto sul parametro selezionato che contiene il 95% della probabilità posteriore. Può essere vagamente pensato come un analogo bayesiano di un intervallo di confidenza. Il parametro TreeHeight fornisce la distribuzione marginale posteriore dell’età della radice dell’intero albero.
Selezionate il parametro TreeHeight e poi fate Ctrl-click su mrcatime(human-chimp) (Command-click su Mac OS X). Questo mostrerà una visualizzazione dell’età della radice e la calibrazione MRCA che abbiamo specificato in precedenza in BEAUti. Potete verificare che la divergenza che abbiamo usato per calibrare l’albero (mrcatime(human-chimp)) ha una distribuzione a posteriori che corrisponde alla distribuzione a priori che abbiamo specificato (Figura 14).
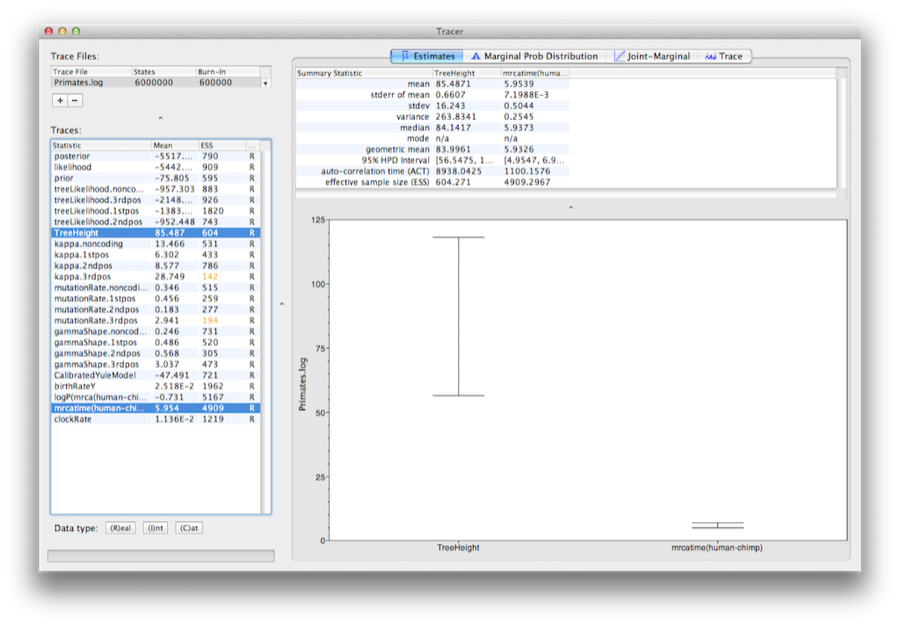
Figura 14: Una schermata degli intervalli HPD al 95% dell’altezza della radice e del MRCA specificato dall’utente (human-chimp) in Tracer.
5 Stime marginali posteriori
Per mostrare i tassi relativi per le quattro partizioni, selezionare il parametro mutationRate per ciascuna delle quattro partizioni e selezionare la scheda densità marginale in Tracer. Il grafico mostra che le posizioni del codone 1 e 2 hanno tassi sostanzialmente diversi (0,456 contro 0,183) ed entrambi sono molto più lenti della posizione del codone 3 con un tasso relativo di 2,941. La partizione non codificante ha un tasso intermedio tra le posizioni codone1 e 2 (0.346). Nell’insieme questo risultato suggerisce una forte selezione purificante sia nelle regioni codificanti che non codificanti dell’allineamento.
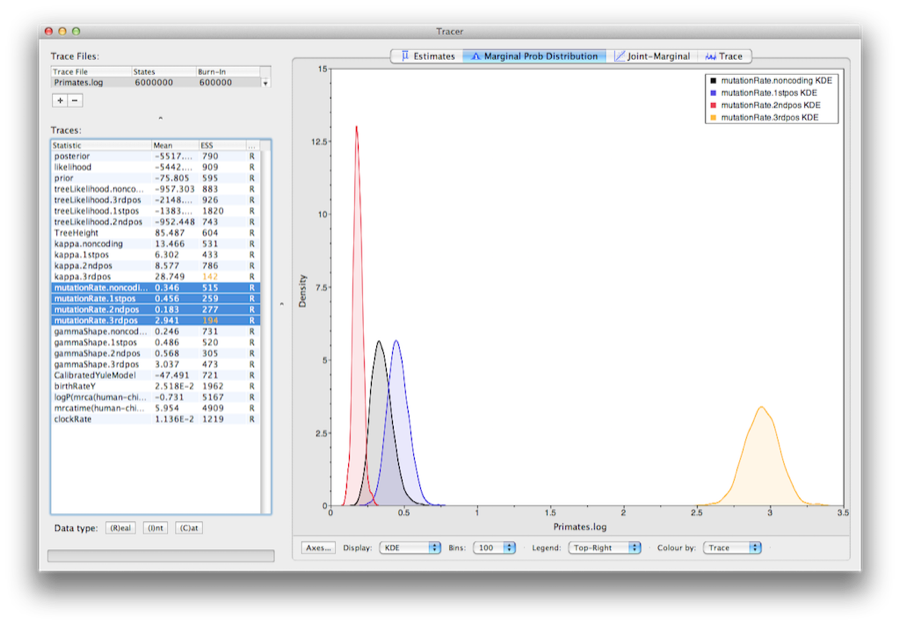
Figura 15: Una schermata delle densità marginali posteriori dei tassi di sostituzione relativi delle quattro partizioni (rispetto al tasso medio ponderato per sito).
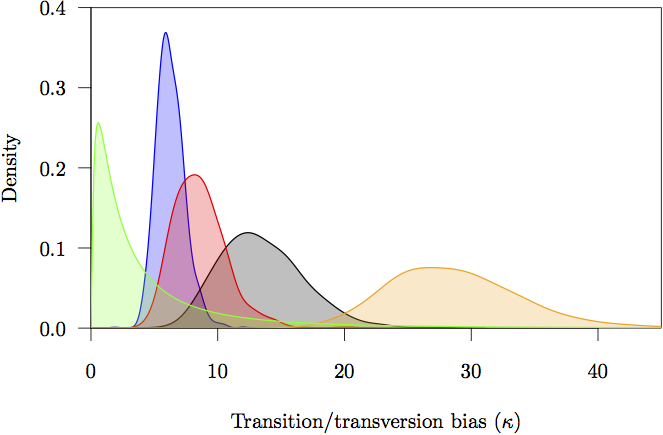
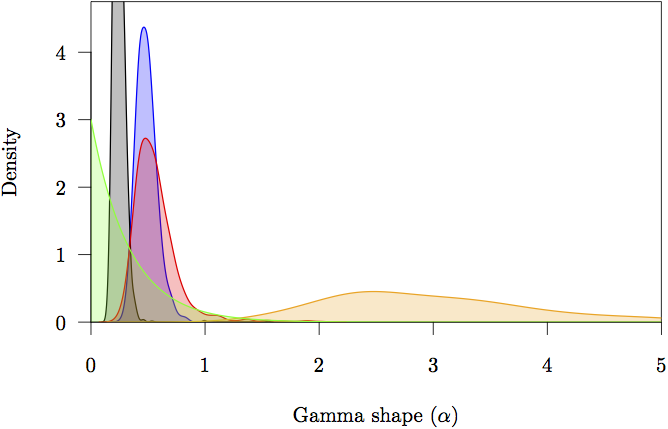
Figura 16: Le densità marginali anteriore e posteriore per i parametri di forma (α). Il priore è in grigio. La stima della densità posteriore per ogni partizione è anche mostrata: non codificante (arancione) e le posizioni del primo (rosso), secondo (verde) e terzo (blu) codone.
Figura 17: Il priore marginale e le densità posteriori per i parametri di bias di transizione/trasversione (κ). Il priore è in grigio. La stima della densità posteriore per ogni partizione è anche mostrata: non codificante (arancione) e posizioni di primo (rosso), secondo (verde) e terzo (blu) codone.
Domande
Qual è il tasso stimato di evoluzione molecolare per questo albero gene (includere l’intervallo HPD 95%)?
Quali fonti di errore include questa stima?
Quanto è vecchia la radice dell’albero (dare la media e l’intervallo HPD 95%)?
6 Ottenere una stima dell’albero filogenetico
BEAST produce anche un campione posteriore di alberi temporali filogenetici insieme al suo campione di stime dei parametri. Questi devono essere riassunti utilizzando il programma TreeAnnotator. Questo prenderà l’insieme degli alberi e troverà quello meglio supportato. Poi annoterà questo albero riassuntivo rappresentativo con le età medie di tutti gli alberi e i corrispondenti intervalli di HPD al 95%. Calcolerà anche la probabilità posteriore di clade per ogni nodo. Eseguite il programma TreeAnnotator e impostatelo come mostrato nella Figura 18.
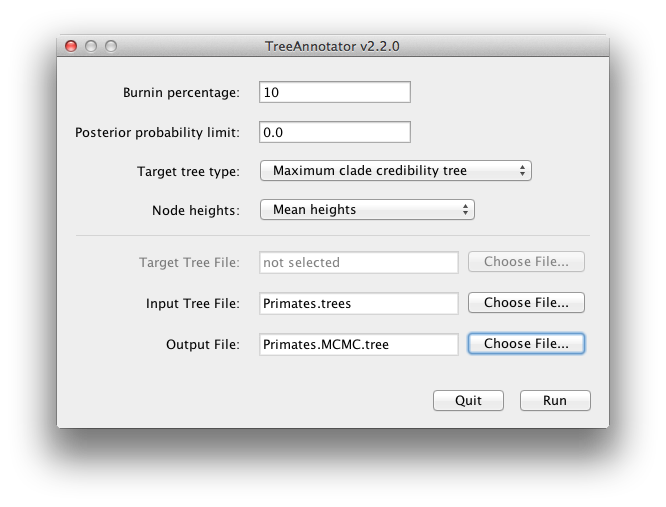
Figura 18: Una schermata di TreeAnnotator.
Il burnin è il numero di alberi da rimuovere dall’inizio del campione. A differenza di Tracer che specifica il numero di passi come burnin, in TreeAnnotator è necessario specificare il numero effettivo di alberi. Per questa esecuzione, avete specificato una lunghezza della catena di 6.000.000 di passi che campionano ogni 1.000 passi. Quindi il file degli alberi conterrà 6.000 alberi e quindi per specificare un burnin del 10% nel campo di testo superiore.
L’opzione limite di probabilità posteriore specifica un limite tale che se un nodo viene trovato a meno di questa frequenza nel campione di alberi (cioè, ha una probabilità posteriore inferiore a questo limite), non verrà annotato. Il valore predefinito di 0,5 significa che solo i nodi visti nella maggioranza degli alberi saranno annotati. Impostatelo a zero per annotare tutti i nodi.
Il tipo di albero di destinazione specifica la topologia dell’albero che sarà annotata. Puoi scegliere un albero specifico da un file o chiedere a TreeAnnotator di trovare un albero nel tuo campione.L’opzione predefinita, Maximum clade credibility tree, trova l’albero con il più alto prodotto della probabilità posteriore di tutti i suoi nodi.
Per le altezze dei nodi, l’impostazione predefinita è Common Ancestor Heights, che calcola l’altezza di un nodo come media del tempo MRCA di tutte le coppie di nodi nel clade. Per gli alberi con grande incertezza nella topologia e quindi molti cladi con basso supporto, alcuni altri metodi possono risultare in alberi con lunghezze di ramo negative. In questa analisi, il supporto per tutte le cladi nell’albero sommario è molto alto, quindi questo non è un problema. Questo imposta le altezze (età) di ogni nodo dell’albero all’altezza media dell’intero campione di alberi per quel clade.
Per il file di input, selezionate il file degli alberi creato da BEAST e selezionate un file per l’output (qui lo abbiamo chiamato Primates.MCC.tree). Ora premi Run e aspetta che il programma finisca.
7 Visualizzare la stima dell’albero
Finalmente, possiamo visualizzare l’albero in un altro programma chiamato FigTree. Esegui questo programma e apri il file Primates.MCC.tree usando il comando Open nel menu File. Ora potete provare a selezionare alcune delle opzioni nel pannello di controllo sulla sinistra. Prima di tutto, spendete l’opzione Trees nel pannello, e spuntate Order nodes e scegliete Ordering by decreasing. Provate a selezionare Node Bars per ottenere le barre di errore dell’età dei nodi. Attivate anche Branch Labels e selezionate posterior per ottenere la visualizzazione della probabilità posteriore per ogni nodo. Se usate un modello di orologio non rigoroso, alla voce Aspetto potete anche dire a FigTree di colorare i rami in base al tasso, e dovreste ottenere qualcosa di simile alla figura 19.
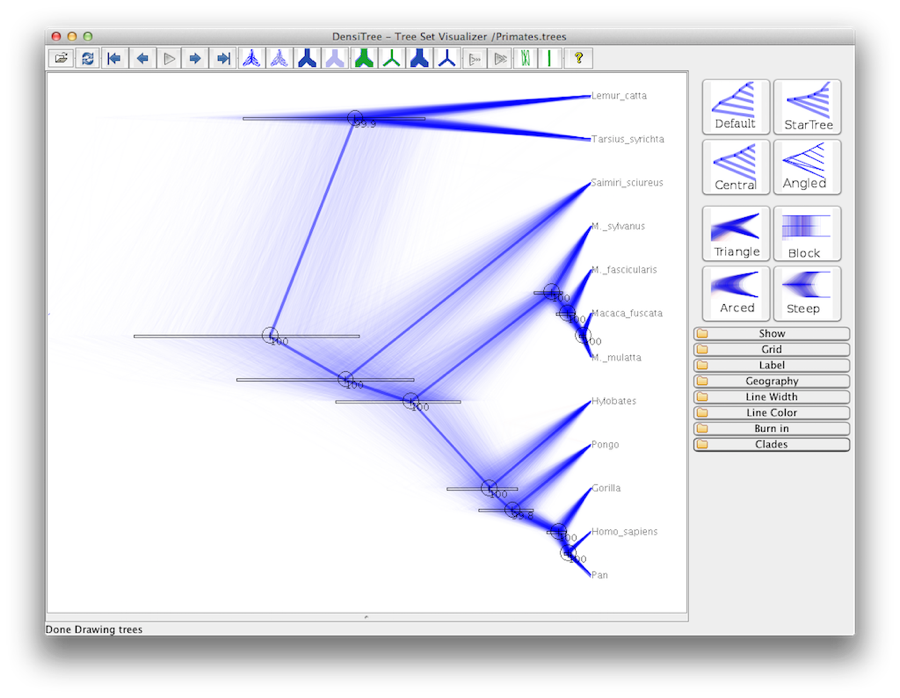
Figura 19: Uno screenshot di FigTree e DensiTree.
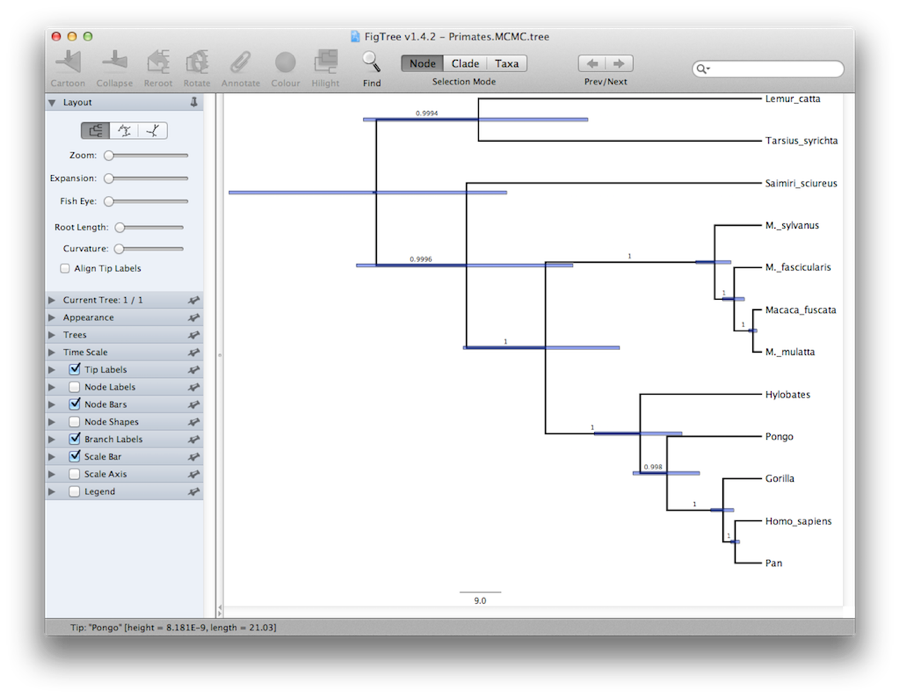
Una visione alternativa dell’albero può essere fatta con DensiTree, che fa parte di Beast 2. Il vantaggio di DensiTree è che è in grado di visualizzare sia l’incertezza nelle altezze dei nodi che l’incertezza nella topologia. Quindi, concludiamo che questa analisi risulta in un consenso molto alto sulla topologia (Figura 19).
Domande
- Il tasso di evoluzione differisce sostanzialmente tra i diversi lignaggi nell’albero?
- DensiTree ha una barra delle cladi (Menu Window/View clade toolbar) per mostrare informazioni sulle cladi.
Qual è il supporto per la clade?
- Puoi sfogliare le topologie in DensiTree usando il menu Browse.La topologia più popolare ha un supporto di oltre il 99%.
Qual è il supporto per la seconda topologia più popolare?
- Nel menu Aiuto, DensiTree mostra alcune informazioni.
Quante topologie ci sono nell’albero?
8 Confrontando i tuoi risultati con il priore
È una buona idea rieseguire l’analisi mentre si campiona dal priore per assicurarsi che le interazioni tra i priori non stiano influenzando le informazioni del priore. L’interazione tra i priori può essere problematica soprattutto quando si usano le calibrazioni, poiché significa mettere più priori sull’albero.
Usando BEAUti, imposta la stessa analisi ma sotto le opzioni MCMC, seleziona l’opzione Sample from prior only. Questo vi permetterà di visualizzare la distribuzione completa dei priori in assenza dei vostri dati di sequenza. Riassumete gli alberi dalla distribuzione a priori completa e confrontate il riassunto con l’albero di riassunto posteriore.
La stima del tempo di divergenza usando la “datazione dei nodi” del tipo descritto in questo capitolo è stata applicata per rispondere a una varietà di domande diverse nell’ecologia e nell’evoluzione. Per esempio, la datazione dei nodi con i fossili è stata usata per determinare la diversità delle specie delle cicadi, per analizzare il tasso di evoluzione nelle piante da fiore e per studiare le origini dei cianobatteri del deserto caldo e freddo.
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Antiche origini determinare globalbiogeography di cianobatteri deserto caldo e freddo, Natura communications2 (2011), 163. Alexei J Drummond e Marc A Suchard, orologi locali casuali bayesiani, o un tasso di governarli tutti, BMC biologia 8 (2010), no. 1, 114. A Gelman, G Roberts, and W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled e Alexei J Drummond, priori dell’albero calibrati per la filogenetica rilassata e la stima del tempo di divergenza, Syst Biol 61 (2012), no. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, e S Mathews, radiazione sincrona recente di un fossile vivente, Scienza 334 (2011), no. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian, e Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), no. 6, 988-993. Stephen A Smith e Michael J Donoghue, tassi di evoluzione molecolare sono collegati alla storia della vita in piante da fiore, scienza 322 (2008), no. 5898, 86-89.
Questo documento è stato tradotto da LATEX daHEVEA.