Incidenza e prevalenza della calcolosi renale continuano ad aumentare nella popolazione generale. Con una prevalenza nell’arco della vita fino al 10%, la nefrolitiasi (NL) e la nefrocalcinosi (NC) sono quindi un peso importante per la salute, soprattutto nel mondo occidentale (1). La NL e la NC sono associate a una significativa morbilità e alla progressione verso la malattia renale cronica a causa delle recidive, dei ripetuti interventi chirurgici/endoscopici e dell’infiammazione concomitante. Ad un livello semplificato, la formazione di calcoli renali risulta da uno squilibrio degli inibitori urinari (ad esempio, citrato, magnesio, uromodulina e pirofosfato) e dei promotori (ad esempio, ossalato, calcio, fosfato, urato e cistina) della cristallizzazione, superando la supersaturazione con conseguente aggregazione, nucleazione e crescita dei calcoli nella placca di Randall (Figura 1). Questo squilibrio può essere dovuto a un’alterata gestione enterale e/o renale di promotori o inibitori, come la malsecrezione enterale di ossalato o il malassorbimento renale di calcio (Figura 1).

Figura 1. Squilibrio degli inibitori urinari e dei promotori della cristallizzazione che porta alla formazione di calcoli renali. La concentrazione degli inibitori e dei promotori urinari è influenzata e controllata da trasportatori intestinali e renali (asse GI – rene). Questi trasportatori e scambiatori, come SLC26A1, sono responsabili della secrezione e dell’assorbimento. Per quanto riguarda l’ossalato, l’iperassorbimento enterale ma la malsecrezione, e/o l’ipersecrezione renale ma il malassorbimento portano a livelli di ossalato urinario che superano la sovrasaturazione e quindi promuovono la cristallizzazione e la successiva formazione di calcoli.
L’eziologia sottostante alla NL è ritenuta multifattoriale con una componente ambientale, alimentare, ormonale e genetica. Negli studi sui gemelli, l’ereditabilità dei calcoli renali è stata stimata al 56% (3), e fino a due terzi dei calcolatori ipercalciurici hanno parenti con NL (4). Anche se i calcoli renali contenenti calcio rappresentano più dell’80% di tutti, la base genetica di tali calcoli rimane in gran parte sconosciuta (5). Ad eccezione delle varianti in CLDN14, TRPV5, SLC34A1, ALPL, CASR, e UMOD, gli studi di associazione genomica non hanno ancora prodotto fattori genetici sostanziali (6-8). Tuttavia, gli alleli di rischio sono stati identificati all’interno di geni che sono stati anche trovati per trasmettere la malattia su base mendeliana, come CASR, SLC34A1 e SLC2A9 (9, 10). Ad oggi, più di 30 singoli geni con un fenotipo definito mendeliano online nell’uomo sono stati identificati per essere implicati nella NL/NC, se mutati (Tabella 1).

Tabella 1. Geni noti per causare forme monogeniche di NL/NC.
Le modalità di eredità nelle forme monogeniche includono la trasmissione autosomica dominante, autosomica recessiva e X-linked. È interessante notare che in molti di questi geni sono stati riportati sia modi di eredità recessivi che dominanti: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 e SLC4A1. Mentre la maggior parte dei disturbi sindromici e congeniti gravi presentano un modello di eredità recessivo (Bartter, Lowe, Dent, FHHNC e acidosi tubulare renale distale con sordità neurosensoriale), le condizioni più lievi sono piuttosto associate a mutazioni nei geni dominanti. La maggior parte delle proteine codificate costituiscono trasportatori renali di soluti (ad esempio, SLC34A1, SLC34A3, e SLC9A3R1), ma anche canali del cloruro (CLCN5), proteine di giunzione stretta (ad esempio, CLDN16/CLDN19), ed enzimi metabolizzanti (ad esempio, AGXT, APRT, e CYP24A1) sono stati trovati difettosi in pazienti con NL/NC. Quindi, il difetto sottostante si trova principalmente nel sistema tubulare del rene stesso e può quindi essere attribuito come tubulopatia. Al contrario, condizioni extrarenali a priori, come nell’iperossaluria primaria (PH) in cui la disfunzione degli enzimi epatici (AGXT, GRHPR e HOGA1) causa un accumulo di ossalato con affezione renale secondaria, sono cause concepibili di NL/NC. Anche se ogni fenotipo di malattia è pensato per rappresentare un’entità relativamente rara, le cause monogeniche possono rappresentare un numero significativo di pazienti grazie alla loro ampia eterogeneità genetica (42). Oltre all’eterogeneità genetica, c’è anche una variazione allelica, dove le varianti troncanti provocano piuttosto una perdita di funzione e le varianti missense (ipomorfe) possono causare difetti piuttosto sottili, che possono essere clinicamente sorvegliati, specialmente nei calcolatori adulti. Un altro fenomeno recentemente apprezzato riguarda gli effetti del dosaggio genico in diversi dei geni della calcolosi renale sopra menzionati. In SLC34A3 per esempio, che codifica uno dei principali trasportatori di fosfato nel tubulo prossimale (NaPiIIc), è stato dimostrato che gli eterozigoti non possono più essere considerati semplicemente come portatori sani, poiché mostrano calcificazioni renali e/o manifestazioni ossee significativamente più frequenti rispetto agli individui wild-type; ma ancora in misura minore rispetto agli individui biallelici (omozigoti e eterozigoti composti) (43). Osservazioni simili sono state riportate per famiglie con mutazioni in CYP24A1 (44). Il contributo dei disordini monogenici alla prevalenza complessiva della calcolosi renale non è stato studiato in modo completo in passato. In particolare, mancano prove genetiche basate su ampi screening di una moltitudine di geni causali in grandi coorti di pazienti. I test genetici completi sono stati troppo costosi e inefficienti in passato. Per la maggior parte degli individui con NL/NC, l’analisi delle mutazioni per un difetto genetico causativo non è stata quindi accessibile, nonostante il fatto che la conoscenza della causa molecolare della NL/NC possa avere importanti conseguenze per la prognosi, la profilassi e/o il trattamento. Solo stime approssimative sono state ricavate da studi di osservazione clinica: sulla base di una vasta raccolta di dati di analisi della composizione dei calcoli, si è concluso che le cause monogeniche non superano il 9,6% nei bambini e l’1,6% negli adulti (45). Nell’ultimo decennio, tuttavia, questa situazione ha cominciato a cambiare, con l’avvento delle tecniche di sequenziamento high-throughput.
Analisi delle mutazioni high-throughput in pazienti con NL/NC
Per studiare i pazienti con calcolosi renale per la presenza di mutazioni patogene nei geni noti della malattia, abbiamo stabilito un pannello genico basato su microfluidica multiplex-PCR e sequenziamento NextGen consecutivo (Fluidigm™/NGS) (46, 47).
In uno “studio pilota”, abbiamo reclutato consecutivamente 268 individui geneticamente irrisolti da cliniche tipiche di calcoli renali; 102 pediatrici e 166 probandi adulti. Come risultato, abbiamo identificato 50 varianti deleterie in 14 dei 30 geni analizzati, portando a una diagnosi molecolare nel 15% dei casi. Nel sottogruppo pediatrico, abbiamo rilevato una mutazione causale nel 21%, mentre tra gli adulti, le varianti deleterie erano presenti nel 11% (Figura 2A) (48). Le mutazioni nel gene della cistinuria SLC7A9 sono state trovate più frequentemente nella coorte degli adulti (Figura 2B). Due studi di follow-up sono stati in grado di confermare questi risultati. In primo luogo, in una coorte esclusivamente pediatrica di 143 pazienti NL/NC, il 17% dei casi è stato spiegato da mutazioni in 14 geni diversi (49). In secondo luogo, in una coorte di 51 famiglie con età di manifestazione della NL/NC prima dei 25 anni, la WES mirata è stata utilizzata per individuare una causa genetica in quasi il 30% (50). Non sorprende che le mutazioni recessive siano state trovate più frequentemente tra i neonati e nei casi di malattia congenita, mentre le condizioni dominanti si sono solitamente manifestate più tardi nella vita. Questi dati indicano che la calcolosi renale genetica è una condizione sottodiagnosticata, nonostante il fatto che la diagnosi molecolare influenzerà potenzialmente la prognosi, la profilassi e/o il trattamento. Una limitazione che vale la pena menzionare, tuttavia, è un potenziale errore di selezione dovuto al reclutamento da cliniche specializzate in calcoli renali in tutti e tre gli studi citati.

Figura 2. Analisi delle mutazioni di 30 geni noti della nefrolitiasi monogenica (NL)/nefrocalcinosi (NC) in 268 pazienti con NL/NC. (A) Frazione di cause monogeniche nella sottocoorte pediatrica e adulta. (B) Numero di cause monogeniche attraverso i geni (rosso denota gli adulti; arancione denota i pazienti pediatrici). Da notare, SLC7A9 è stato trovato più frequentemente mutato, soprattutto nei giovani adulti.
Identificazione di nuovi geni di malattia umana da approccio del gene candidato
L’analisi di mutazione ad alta capacità è anche usato per lo screening di varianti patogene in vari geni candidati. Uno dei risultati recenti più interessanti è stata la scoperta di mutazioni umane in SLC26A1 (32). Dalla prima descrizione della formazione di calcoli renali di Ca-ossalato (CaOx) e NC in topi Slc26a1 (Sat1)-knockout da Dawson et al. nel 2010, SLC26A1 è stato un gene candidato bona fide NL (51). SLC26A1 codifica uno scambiatore di anioni espresso sulla membrana basolaterale dei tubuli renali prossimali, ileo e digiuno. Di conseguenza, utilizzando un approccio candidate-gene, sono state identificate varianti patogene in esseri umani con una storia di insorgenza precoce di CaOx-NL, ovvero due individui non imparentati con varianti missense bialleliche (32). Funzionalmente, la patogenicità delle varianti identificate è stata dimostrata in vitro, portando ad un mis-trafficking intracellulare e ad un’alterata attività di trasporto (32). Il difetto di SLC26A1 costituisce quindi una nuova causa di CaOx-NL e dovrebbe essere considerato quando si esaminano gli individui per le cause di formazione ricorrente di calcoli di CaOx.
Nuovo registro clinico dei pazienti per la calcolosi renale ereditaria
La maggior parte dei dati epidemiologici sull’aumento della prevalenza nei paesi occidentali deriva dai database statunitensi. Anche se urgentemente necessari, i database europei centralizzati non sono disponibili al momento. Dato che i suddetti studi genetici sulla prevalenza della calcolosi renale ereditaria sono stati eseguiti con piccole coorti di centri specializzati sia in Europa che negli Stati Uniti, una traduzione alla situazione generale in Europa non è valida. Mentre negli Stati Uniti, il Rare Kidney Stone Consortium costituisce una piattaforma che integra e coordina il registro, la scienza di base e le attività di ricerca clinica per condizioni rare come la cistinuria, il PH, il deficit di APRT, la malattia di Dent e Lowe, nessuna raccolta di dati comparabile sui pazienti con malattia renale ereditaria dei calcoli è stata implementata né in Europa né in Germania oggi. In collaborazione con l’esistente registro europeo PH, OxalEurope (Prof. Bernd Hoppe, Università di Bonn), e grazie al finanziamento della Deutsche Forschungsgemeinschaft e della Else Kröner-Fresenius Stiftung, abbiamo recentemente istituito un “Registro dei pazienti clinici per la calcolosi renale ereditaria” all’Università di Lipsia. Il registro è sostenuto a livello nazionale dalle società tedesche di nefrologia degli adulti (DGfN) e di nefrologia pediatrica (GPN). È inoltre iscritto al Registro tedesco degli studi clinici (DRKS-ID: DRKS00012891). Come parte fondamentale del reclutamento dello studio, l’analisi delle mutazioni ad alta produttività per i geni noti e nuovi della calcolosi renale è offerta su una base di ricerca per i pazienti senza una diagnosi molecolare stabilita ma con un quadro clinico che indica una suscettibilità genetica sottostante: ad esempio, età precoce di insorgenza (<40 anni), storia familiare positiva, fenotipi indicativi come NC, cistinuria, o RTA, e NL gravemente ricorrente (>3×) (Tabella 2). Mentre i pazienti con una diagnosi genetica già stabilita vengono generalmente arruolati, i casi con cause secondarie di NL/NC, come la malignità, la sarcoidosi e l’iperparatiroidismo primario, non vengono inclusi nell’analisi genetica. Per arruolare attivamente i pazienti, un centro clinico avrà solitamente bisogno dell’approvazione da parte dell’Institutional Review Board locale; un processo per il quale offriamo il nostro aiuto e assistenza fornendo i rispettivi modelli. Dopo l’approvazione etica, il modulo di consenso e i fogli di dati clinici (ad esempio, il questionario clinico) possono essere scaricati dal nostro sito web del registro (http://www.mks-registry.net). Per garantire un’accurata fenotipizzazione clinica, chiederemo informazioni sostanziali sul paziente come etnia, consanguineità, storia familiare, età di insorgenza, recidiva (definita come ogni evento putativo di calcoli renali), assunzione giornaliera di liquidi, interventi chirurgici e coinvolgimento extrarenale, tra gli altri. I documenti possono essere compilati dal paziente con l’aiuto del medico arruolatore. Inoltre, i parametri biochimici del siero, tra cui creatinina, eGFR, PTH, vitamina D, elettroliti, acido urico, e l’analisi delle urine (pH, calcio, fosfato, magnesio, acido urico, citrato, ossalato, e cistina, preferibilmente dalle urine delle 24 ore, se non dalle urine spot), così come i dati sull’analisi della composizione dei calcoli saranno richiesti al momento dell’arruolamento. Tenendo conto del fatto che la raccolta delle urine a 24 ore e l’analisi della composizione dei calcoli non vengono eseguite di routine in tutte le istituzioni, includiamo questi parametri se disponibili. Le visite di follow-up clinico a 2 anni dei pazienti arruolati sono auspicabili ma non obbligatorie. Dopo la registrazione, i centri clinici che reclutano riceveranno un login personalizzato per inserire i dati dei pazienti tramite il nostro sito web del registro (http://www.mks-registry.net). In alternativa, offriamo la possibilità di inserire i dati elettronicamente quando ci vengono inviati su carta. I dati inseriti saranno memorizzati su un server protetto e vi potranno accedere i centri clinici partecipanti per visualizzare i dati dei loro pazienti. I seguenti siti web forniscono ulteriori informazioni:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
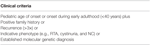
Tabella 2. Criteri di inclusione per l’analisi delle mutazioni nel registro dei pazienti clinici.
In sintesi, la malattia dei calcoli renali è una condizione sempre più prevalente che è clinicamente eterogenea e poco compresa, in particolare i suoi driver genetici. Come indicato da una serie di studi recenti, le condizioni monogeniche sono molto probabilmente sottostimate nella prevalenza. Con l’implementazione di un registro centralizzato dei pazienti sulla calcolosi renale ereditaria, contribuiremo a superare, almeno in parte, la vasta lacuna di conoscenze sulla genetica della calcolosi renale. In questo contesto, i registri clinici sono fonti preziose per diverse ragioni: in primo luogo, delineare meglio le associazioni fenotipo-genotipo sarà fondamentale per una più precisa stratificazione dei pazienti nei futuri studi di ricerca clinica. In secondo luogo, l’identificazione di nuovi geni della malattia con nuovi meccanismi di malattia diminuirà la lacuna dell’eziologia sconosciuta della NL/NC; e in terzo luogo, la decifrazione di nuovi bersagli molecolari aiuta ad aprire la strada allo sviluppo di farmaci di prevenzione delle recidive nelle famiglie gravemente colpite.
Dichiarazione etica
Questo studio è stato condotto in conformità alle raccomandazioni della “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” con consenso informato scritto da tutti i soggetti. Tutti i soggetti hanno dato il consenso informato scritto in conformità con la Dichiarazione di Helsinki. Il protocollo è stato approvato dalla “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig.”
Contributi degli autori
JH ha concepito e scritto il manoscritto. BH, AS, LM e RS hanno curato il manoscritto e costruito l’infrastruttura del registro che viene presentata al lettore.
Dichiarazione di conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Finanziamento
La ricerca è finanziata da sovvenzioni di progetto da DFG (HA 6908/2-1) e EKFS (2016_A52) a JH. Questo lavoro è stato ulteriormente sostenuto dal Ministero federale dell’istruzione e della ricerca (BMBF), Germania, FKZ: 01EO1501 a JH.
1. Scales CD Jr., Smith AC, Hanley JM, Saigal CS. Prevalenza di calcoli renali negli Stati Uniti. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Genetica della nefrolitiasi ipercalciurica: malattia della pietra renale. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Progressi nella comprensione della genetica della nefrolitiasi contenente calcio. J Am Soc Nephrol (2017) 28: 748-59. doi:10.1681/ASN.2016050576
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. varianti di sequenza nel gene CLDN14 associano con calcoli renali e densità minerale ossea. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Associazione di varianti a UMOD con malattia renale cronica e rene pietre-ruolo di età e malattie comorbide. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Varianti comuni e rare associate a calcoli renali e tratti biochimici. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 influenza le concentrazioni di acido urico con pronunciati effetti sesso-specifici. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutazioni nel trasportatore di glucosio 9 gene SLC2A9 causare ipouricemia renale. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identificazione e caratterizzazione di un gene con sostituzioni di base associate al fenotipo ipercalciuria assorbente e bassa densità ossea spinale. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting di peroxisomal L-alanina: glyoxylate aminotransferase ai mitocondri in pazienti iperossaluria primaria dipende dall’attivazione di una sequenza criptica di targeting mitocondriale da una mutazione puntiforme. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Adenina fosforibosiltransferasi umana. Identificazione di mutazioni alleliche a livello nucleotidico come causa di carenza completa dell’enzima. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutazioni in ATP6N1B, che codifica un nuovo rene vacuolare pompa protonica 116-kD subunità, causa recessiva distale acidosi tubulare renale con udito conservato. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutazioni nel gene che codifica B1 subunità di H +-ATPasi causare acidosi tubulare renale con sordità neurosensoriale. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. La sindrome da carenza di anidrasi carbonica II in una famiglia belga è causata da una mutazione puntiforme a un residuo invariante di istidina (107 His→Tyr): struttura completa del normale gene umano CA II. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. Una sindrome familiare di ipocalcemia con ipercalciuria dovuta a mutazioni nel recettore calcio-sensing. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. Una base molecolare comune per tre malattie ereditarie dei calcoli renali. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutazioni nel gene del canale del cloruro, CLCNKB, causa sindrome di Bartter tipo III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, una proteina di giunzione stretta renale necessaria per il riassorbimento paracellulare di Mg2+. Scienza (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutazioni nella stretta giunzione gene claudin 19 (CLDN19) sono associati con spreco di magnesio renale, insufficienza renale e grave coinvolgimento oculare. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutazioni in CYP24A1 e ipercalcemia infantile idiopatica. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nefrocalcinosi (sindrome renale smalto) causata da mutazioni autosomiche recessive FAM20A. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Il gene che codifica l’idrossipiruvato reduttasi (GRHPR) è mutato in pazienti con iperossaluria primaria di tipo II. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. La mutazione HNF4A R76W causa la sindrome di Fanconi dominante atipica oltre a un fenotipo delle cellule β. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. mutazioni in DHDPSL sono responsabili per iperossaluria primaria di tipo III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identificazione di 17 mutazioni indipendenti responsabili del deficit di ipoxantina-guanina fosforibosiltransferasi umana (HPRT). Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. eterogeneità genetica della sindrome di Bartter rivelato da mutazioni nel canale K +, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transitorio sindrome di Bartter prenatale e mutazioni MAGED2. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Marcatori fiancheggiatori strettamente collegati per la sindrome oculocerebrorenale di Lowe, con applicazione alla valutazione del portatore. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. La sindrome di Bartter, alcalosi ipokaliemica con ipercalciuria, è causata da mutazioni nel Na-K-2Cl cotrasportatore NKCC2. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutazioni in SLC26A1 causare nefrolitiasi. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Identificazione molecolare di uno scambiatore di anioni di urato renale che regola i livelli di urato nel sangue. Natura (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nefrolitiasi e osteoporosi associate a ipofosfatemia causata da mutazioni nel cotrasportatore sodio-fosfato tipo 2a. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. rachitismo ereditario ipofosfatemico con ipercalciuria è causato da mutazioni nel sodio-fosfato cotrasportatore gene SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cistinuria causato da mutazioni in rBAT, un gene coinvolto nel trasporto di cistina. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. L’acidosi tubulare renale distale familiare è associata a mutazioni nel gene dello scambiatore di anioni delle cellule rosse (banda 3, AE1). J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Cistinuria non tipo I causata da mutazioni in SLC7A9, che codifica una subunità (bo,+AT) di rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. Mutazioni NHERF1 e reattività dell’ormone paratiroideo renale. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Prove suggestive per un gene di suscettibilità vicino al recettore della vitamina D nella formazione di calcoli idiopatici. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identificazione di due mutazioni nel gene della xantina deidrogenasi umana responsabile del classico tipo I xantinuria. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Hildebrandt F. Malattie renali genetiche. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutazioni in SLC34A3/NPT2c sono associati con calcoli renali e nefrocalcinosi. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Fenotipi clinici e biochimici di adulti con mutazioni monoalleliche e bialleliche CYP24A1: prove di effetto di dose del gene. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Recenti acquisizioni diagnostiche e terapeutiche. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput analisi delle mutazioni in pazienti con una ciliopatia associata a nefronoftisi applicando multiplexed array basato su PCR amplificazione barcoded e next-generation sequencing. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identificazione di 99 nuove mutazioni in una coorte mondiale di 1.056 pazienti con una ciliopatia nefroftisica. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Quattordici geni monogenici rappresentano il 15% della nefrolitiasi/nefrocalcinosi. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalenza di cause monogeniche in pazienti pediatrici con nefrolitiasi o nefrocalcinosi. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Il sequenziamento dell’esoma intero rileva frequentemente una causa monogenica nella nefrolitiasi ad esordio precoce e nella nefrocalcinosi. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolitiasi ed epatotossicità sono legati al trasportatore di anioni Sat1 nei topi. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar