Alexei Drummond, Andrew Rambaut, Remco Bouckaert, and Walter Xie
1 Introduction
このチュートリアルではベイズ進化解析ソフトBEASTについて簡単に紹介する。 このチュートリアルでは、化石証拠からの較正情報の存在下で、遺伝子系統と関連する分岐時間の共推定を行う。
以下のソフトウェアが必要です。
- BEAST – このパッケージには BEAST プログラム、BEAUti、TreeAnnotator およびその他のユーティリティ プログラムが含まれています。 このチュートリアルは、複数のパーティションをサポートするBEAST v2.2.x用に書かれています。 http://www.beast2.org/.
- Tracer – このプログラムはBEAST(および他のベイズMCMCプログラム)の出力を探索するために使用されます。 連続パラメータの分布をグラフィカルかつ定量的に要約し、診断情報を提供します。 3586>http://tree.bio.ed.ac.uk/software/.
- FigTree – 分子系統樹、特にBEASTを使って得られた系統樹を表示・印刷するためのアプリケーションです。 この記事の執筆時点では、現在のバージョンはv1.4.2です。 http://tree.bio.ed.ac.uk/software/.
このチュートリアルでは、12種の霊長類から採取した配列のアライメントの解析について説明します(図1参照)。 目標は系統、各系統の進化率、未校正の分岐の年代を推定することです。
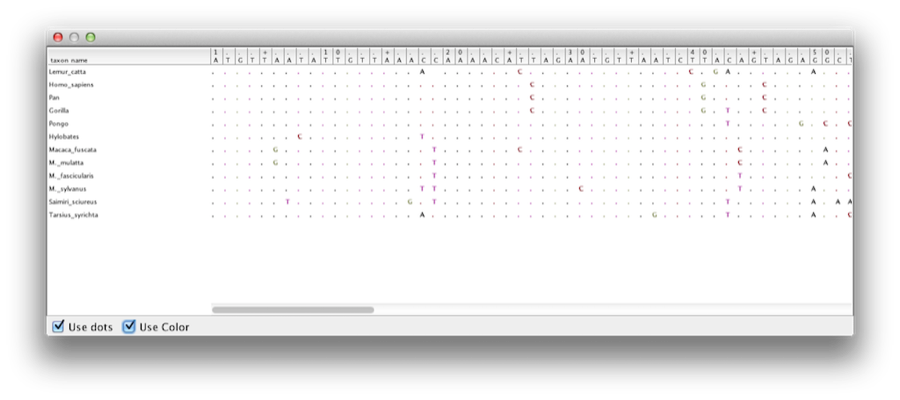
Figure 1: Alignment for primates.
最初のステップはDATAまたはCHARACTERSブロックがあるNEXUSファイルをBEAST XML入力ファイルに変換することである。 これは、BEAUti(Bayesian Evolutionary Analysis Utilityの略)というプログラムを使って行います。 これは進化モデルやMCMC解析のオプションを設定するためのユーザーフレンドリーなプログラムです。 次に、BEAUTiが生成した入力ファイル(データ、モデル、解析設定が含まれる)を使って、実際にBEASTを実行します。
2 BEAUti
プログラムBEAUtiは、BEASTのモデルパラメータを設定するためのユーザーフレンドリーなプログラムである。 BEAUtiのアイコンをダブルクリックして、BEAUtiを実行します。 BEAUtiは一度起動すれば、どのコンピュータシステムで実行しても同じように表示されます。 このチュートリアルでは、Mac OS X版を使用していますが、LinuxやWindows版も同じレイアウトと機能です。
2.1 NEXUSファイルのロード
NEXUSフォーマットのアライメントをロードするには、ファイルメニューからインポートアライメント…を選択するか、ファイルをPartitionsパネルの真ん中にドラッグしてください。
primate-mtDNA.nex というサンプルファイルは、Mac と Linux では examples/nexus/ ディレクトリから、Windows では BEAST をインストールしたディレクトリの examples/nexus/ ディレクトリから入手できます。
関連パッケージがインストールされていれば、Add Partitionウィンドウ(図2)がポップアップ表示されます。 純粋な “BEAST 2 “を使用している場合は、次の段落に進むことができます。 そうでない場合は、[Add Alignment] を選択して [OK] をクリックして続行します。
Figure 2: Add Partition window (Only if related packages are installed).
partitions でコードの重複があると警告メッセージウィンドウ (Figure 3) が表示されます。
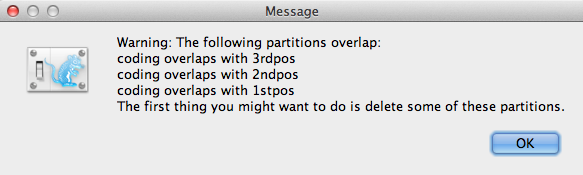
Figure 3: Warning message window (Only if there is any coding overlaps in the partitions).
ロードするとメインパネルに5文字のパーティションが表示されます(Figure 4)。 アライメントはタンパク質のコード部分と非コード部分に分けられ、コード部分はコドン位置1、2、3で分割されている。 coding’ パーティションは ‘1stpos’, ‘2ndpos’, ‘3rdpos’ パーティションと同じヌクレオチドを参照しているため、次のステップに進む前に削除する必要があります。 coding’パーティションを削除するには、行を選択し、テーブルの下部にある’-‘ボタンをクリックします。
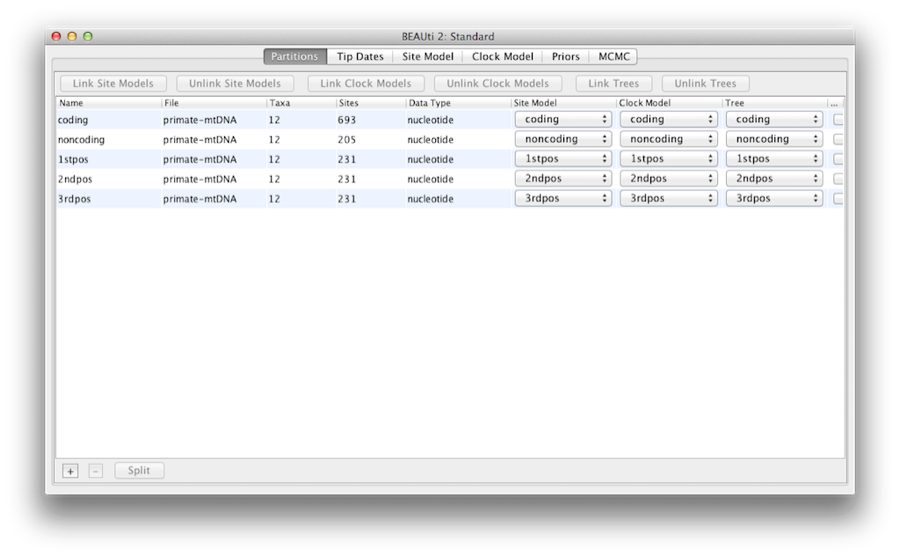
図4:BEAUtiのデータタブのスクリーンショットです。
パーティションモデルのリンク/リンク解除
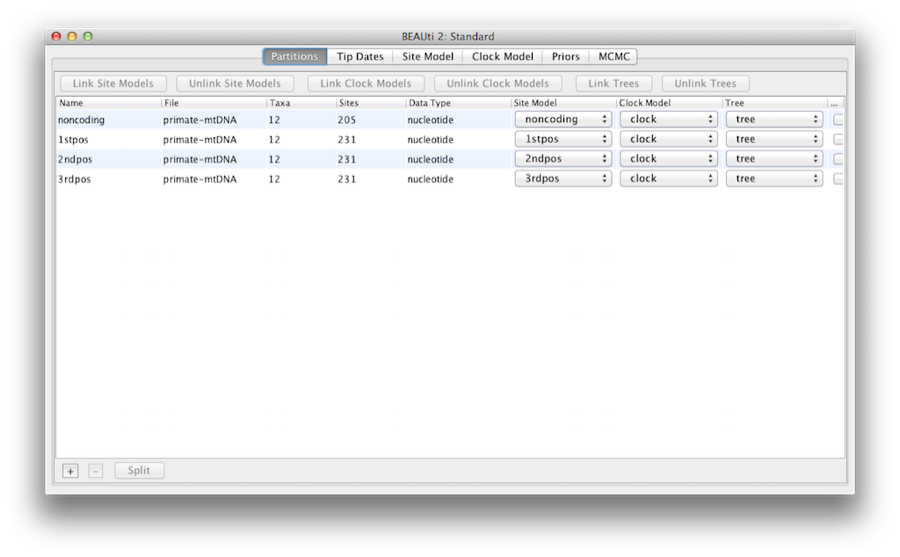
Figure 5: 時計モデルおよびツリーをリンクして名前を変更した後のBEAUtiの「パーティション」タブのスクリーンショットです。
配列はリンクされているので(つまり、鳥や哺乳類で組換えを起こさないと考えられているミトコンドリアゲノムからすべて)、同じ祖先を共有し、モデル内のタイムツリーを共有するはずである。 簡単のために、各分岐の進化速度も同じとし、同じ「時計モデル」とする。速度不均質性のモデル化は各分岐内の部位間不均質性に限定し、各分岐が異なる平均進化速度を持つことも認めることにする。
そこで、この時点でクロックモデルとツリーをリンクさせる必要があります。 パーティションパネルで、表中の4つのパーティションをすべて選択し(またはなし、デフォルトではすべてのパーティションが影響を受けます)、Link Treesボタンをクリックし、次にLink Clock Modelsボタンをクリックします(図5を参照ください)。 次に、Clock Model列の最初のドロップダウンメニューをクリックし、共有クロックモデルの名前を’clock’に変更します。 同様に、共有ツリーの名前も’tree’に変更します。 これにより、以下のオプションや生成されるログファイルが読みやすくなります。
2.2 置換モデルの設定
次のステップは、置換モデルの設定です。 次に、メインウィンドウの上部にあるSite Modelsタブを選択します(すべての分類群が同時期のサンプルであるため、Tip Datesタブはスキップします)。 これにより、BEAST の進化モデルの設定が表示されます。 使用できるオプションは、データがヌクレオチドかアミノ酸か、バイナリデータか、一般的なデータかによって異なります。 霊長類のヌクレオチドアライメントを読み込んだ後に表示される設定は、ヌクレオチドデータのデフォルト値となりますので、いくつか変更する必要があります。
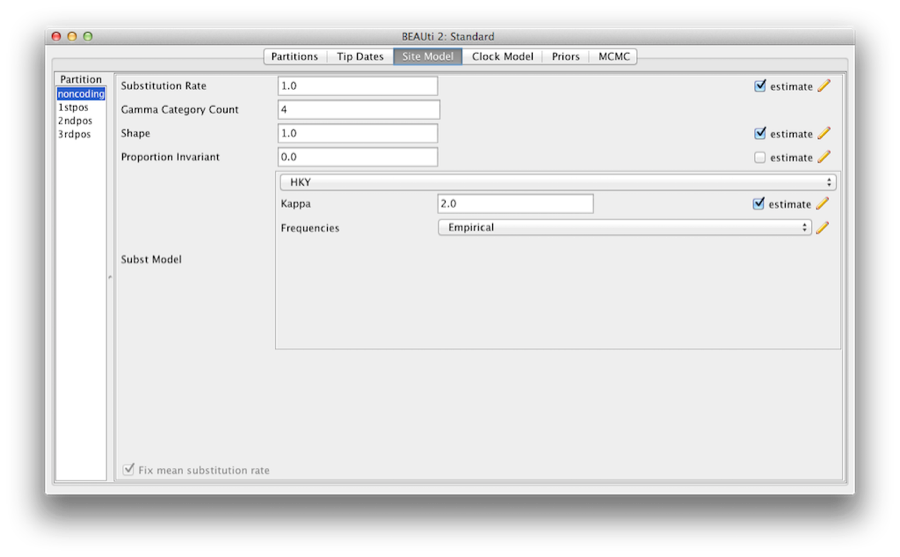
Figure 6: BEAUtiのサイトモデルタブのスクリーンショット。
モデルの大部分は見覚えがあるはずである。 まず、Gamma Category Countを4に設定し、Shapeパラメータの’estimate’にチェックを入れる。 これは、各パーティションのサイト間のレート変動をモデル化することを可能にします。 4から6のカテゴリーは、ほとんどのデータセットで十分に機能しますが、より多くのカテゴリーを持つことは、ほとんど追加的な利益のために計算に時間がかかることに注意してください。 Proportion Invariantの項目は0に設定しておく。
次に、Subst ModelのドロップダウンメニューからHKYを選択する。 理想的には、置換モデルは各パーティションでデータに最も適合するものが選択されるべきですが、ここでは簡単のために、すべてのパーティションでHKYを使用します。 さらに、Frequencies ドロップダウン・メニューから Empirical を選択します。 これにより、頻度がデータで観測された割合に固定されます(サイトモデルのリンクを解除した後、各パーティションごとに)。 このアプローチは、これらのパラメータを明示的に推定することなく、データへの良いフィットを得ることができることを意味します。 ここでは、単にログファイルを少し短くして、この演習の後の部分で読みやすくするために、この方法をとります。
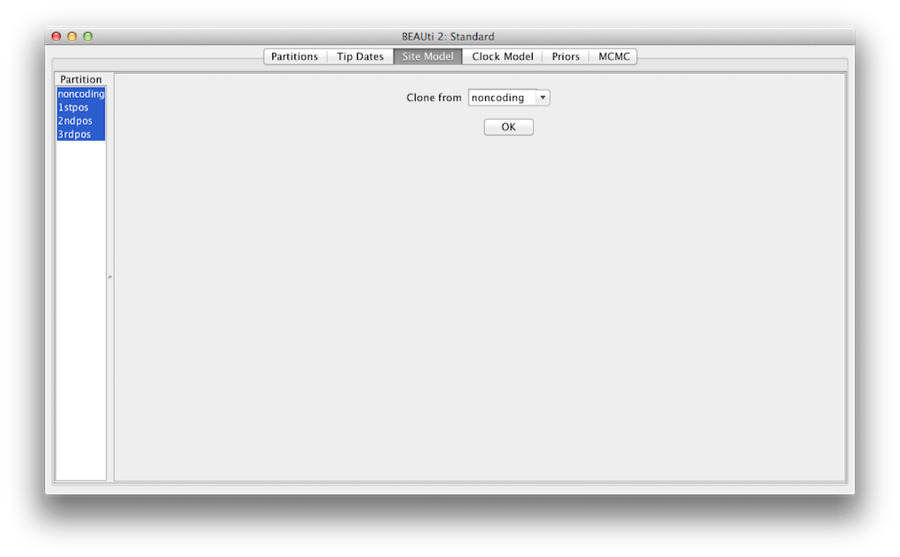
Figure 7: Clone configuration from one site model to others.
最後に Substitution rate parameter の ‘estimate’ ボックスをチェックして Fix mean mutation rate チェックボックスを選択します。
最後に、「shift」キーを押しながら左側のサイトモデルをすべて選択し、「OK」をクリックしてノンコーディングから1stpos、2ndpos、3rdposに設定をクローンします(図7)。 各サイトモデルを確認すると、ご覧のように同じ構成になっています。
2.3 時計モデルの設定
次のステップは、メインウィンドウの上部にあるClock Modelsタブを選択することです。 ここで分子時計モデルを選択します。 この演習では、このデータは非常に時計的であり、モデルに含まれる枝の間の速度変化を必要としないので、デフォルト値の厳密な分子時計のまま選択することにする。
時計らしさをテストするには、(i)緩和された時計モデルで解析を実行し、データによってどの程度レートの変動が暗示されるかをチェックする(これについては変動係数を参照)、(ii) パスサンプリングを使用して厳格な時計と緩和な時計のモデル比較を行う、または (iii) 木における各枝にそれ自身の分岐率が必要かどうかを明確に考慮したランダム局所時計モデルを使用する、が考えられます。
2.4 Priors
Priorsタブでは、モデル内の各パラメータに対してプライヤーを指定することができます。 サイトモデルとクロックモデルタブでモデルを選択すると、モデルに様々なパラメータが含まれることになり、これらはプライヤータブに表示されます(図8参照)。
ここで、樹木事前分布としてCalibrated Yuleモデルを使用したいことも指定します。 Yuleモデルは、種分化の単純なモデルで、異なる種からの配列を考慮するときに一般的により適切です。
2.4.1 キャリブレーション・ノードの定義
ここで、我々の化石知識に基づいて、キャリブレーション・ノード上の事前分布を指定する必要があります。 これは樹木の較正として知られている。 追加の事前分布を定義するには、事前分布のリストの下にある小さな+ボタンを押します。 もし、このボタンが表示されていない場合は、パネルを一番下までスクロールして、+ボタンを見つけてください。 すると、系統樹の分類群のサブセットを定義するためのダイアログが表示されます。 一度分類群セットを作成すると、後でその直近の共通祖先(MRCA)のキャリブレーション情報を追加することができるようになります。
分類群セットラベルの項目を埋めて、分類群セットに名前を付けます。 ホモ・サピエンスとパンの分類を含むので、human-chimp と呼びます。 下のリストに、利用可能な分類群が表示されます。 2つの分類群を順番に選択し、>>矢印ボタンを押します。 (これはCalibrated Yule事前分布と組み合わせて使用する較正ノードであるため、単系統を強制する必要があり、Monophyleticと書かれたチェックボックスを選択します(図9)。 これは、MCMC分析の過程で、ヒトとチンパンジーのグループ分けが単系統に保たれるように、木のトポロジーを制約します。
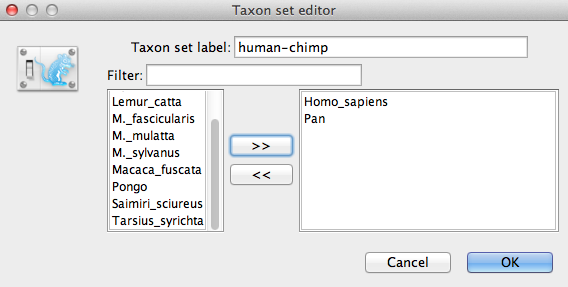
図9:BEAUtiのタクソンセットエディター
キャリブレーション情報をエンコードするために、ヒト-チンパンジーのMRCAの分布を指定する必要があります。新しく追加したヒト-チンパンジー.priorの右側のドロップダウンメニューから対数正規分布を選択します。 黒い三角形をクリックすると、確率密度関数のグラフと対数正規分布のパラメータが表示されます。 これにより、95%の確率で5-7Maの範囲をカバーすることになる。 これは、ヒトとチンパンジーの最も最近の共通祖先の日付の現在のコンセンサス推定にほぼ対応する(図10)。
Priorsパネルに表示されている事前分布が、本当にモデルのパラメータに関する事前情報を反映していることを確信する必要があります。 最後に、全体の分子時計速度(clockRate)と種分化率(birthRateY)についての、拡散的な「非情報的」だが適切な事前分布をユールツリーの事前分布で指定することにします。 それぞれについて、ドロップダウンメニューからGammaを選択し、矢印ボタンを使って、Gamma事前分布のパラメータを明らかにするためにビューを拡張してください。 クロックレートとユール出生率の両方について、Alpha(shape)パラメータを0.001に、Beta(scale)パラメータを1000に設定します(図11)
デフォルトでは、それぞれのガンマ形状パラメータは、平均を1とする指数優先分布を持っています。 HKYモデルのカッパパラメータは対数正規分布(1,1.25)であり、これは遷移/転回バイアスの現実的な値の範囲に関する経験則とほぼ一致しています。
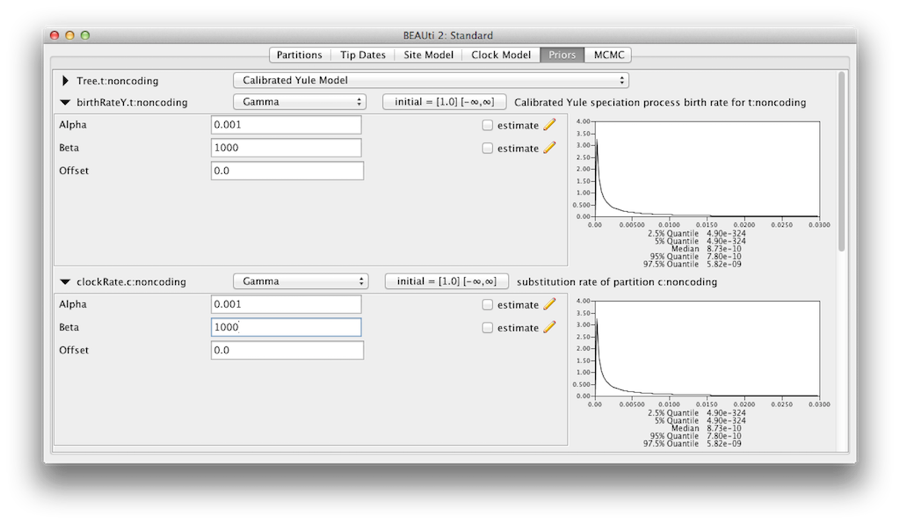
Figure 11: Gamma prior.
2.5 MCMCオプション設定
次のタブ、MCMCでは、MCMC実行時間やファイル名を制御するより一般的な設定を提供します。
まず、Chain Length(連鎖の長さ)です。 これは、MCMCが終了する前に鎖で行うステップの数です。 どの程度の長さにするかは、データセットのサイズ、モデルの複雑さ、必要な答えの質によって決まります。 デフォルトの10,000,000は完全に任意であり、データセットの大きさに応じて調整する必要があります。 このデータセットでは、鎖長を 6,000,000 に設定します。これは、ほとんどのモダンなコンピューターで適度に速く実行されます (数分)。
Store Every フィールドは、状態がファイルに保存される頻度を決定します。 状態を定期的に保存することは、コンピュータ環境があまり信頼できず、BEAST の実行が中断される可能性があるような場合に便利です。 Pre Burninフィールドは、解析の一番最初に記録されないサンプル数を指定します。 Store EveryとPre Burninフィールドはデフォルト値のままにしておきます。 これらの下にログファイルの詳細が表示されます。
次のオプションは、Markovchainのパラメータ値が画面に表示され、ログファイルに記録される頻度を指定します。画面出力は、単にプログラムの進行状況を監視するためのもので、任意の値を設定できます(ただし、あまり小さく設定すると、画面に表示される情報量が多すぎて、かえってプログラムの速度が低下します)。 ログファイルについては、チェーンの全長と比較して値を設定する必要があります。 サンプリングの頻度が高すぎると、ファイルが非常に大きくなり、解析の正確さの点ではあまり利点がありません。 サンプリング頻度が低すぎると、ログファイルにはパラメータの分布に関する十分な情報が記録されません。 この演習では、trace logとtree logの頻度を1,000に、screen logを10,000に設定し、trace logファイルのファイル名としてPrimates.log、tree logファイルのファイル名としてPrimates.treesを指定します。
- Windows OSを使用している場合、Windowsがこれらをテキストファイルとして認識するように、これらのファイル(Primates.log.txtとPrimates.trees.txt)に拡張子.txtを追加することをお勧めします。
2.6 BEAST XML ファイルの生成
BEAST XML ファイルを作成する準備が整いました。 これを行うには、[ファイル]メニューから[保存]オプションを選択します。 デフォルトのプライヤーをチェックし、適切な名前で保存します(通常、ファイル名は.xmlで終わります。
3 BEASTの実行
Figure 12: BEASTのスクリーンショット
ここでBEASTを実行して入力ファイルを求められたら、新しく作ったXMLファイルを入力として提供しましょう。 BEAST は、画面に情報を報告し終わるまで実行されます。 実際の結果ファイルは、入力ファイルと同じ場所にディスクに保存されます。 画面への出力は次のようなものになります。
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds最初にアラインメントとどの樹木尤度を使用したかに関する有用な情報があることに注意されたい。 また、解析に関連するすべての引用文献が実行の最初に記載されており、解析について報告する原稿に簡単にコピーすることができる。
最後に演算子解析が出力され、解析に使用されたすべての演算子と、その演算子が試され、受け入れられ、拒否された頻度(それぞれ #total, #accept, #reject 列を参照)が一覧表示されます。 受容率とは、あるオペレータが提案のために選択されたとき、受容された回数の割合のことです。 一般に、採択率が高く、例えば0.5以上であれば、提案は保守的であり、パラメータ空間を効率的に探索できていないことを示します。 一方、受理率が低い場合は、プロポーザルが積極的すぎることを示し、ほとんどの場合、事後値が低いために拒否される状態になります。 BEASTに実装されている多くの(全てではありませんが)演算子では、0.234の受け入れ率が目標値です。 最終的な合格率が目標値に近くない場合、BEAST は調整パラメータの新しい値を提案し、この値は演算子解析に出力されます。 この場合、チューニングパラメータを持つ演算子では、すべての合格率が良好となります。 チューニングパラメータを持たない演算子には、この分析のためのwideexchange演算子およびWilson-Balding演算子が含まれます。 これらの演算子はいずれも大きなステップで木のトポロジーを変えようとするが、データは圧倒的に単一のトポロジーを支持するので、これらの過激な提案はほとんど拒否される。
4 結果を分析する
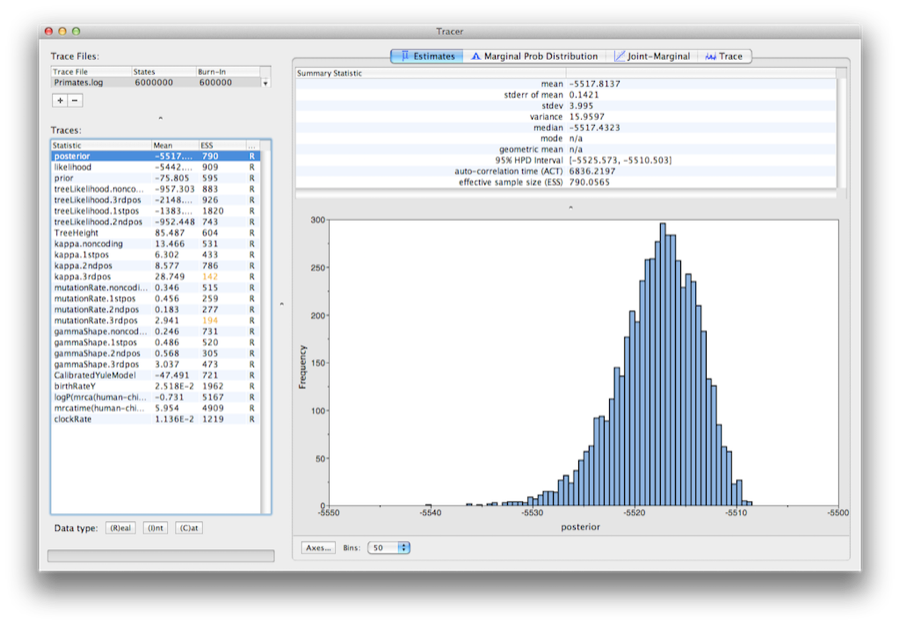
Figure 13: Tracer v1.6 のスクリーンショット
BEASTの出力を分析するには、トレーサーというプログラムを実行します。 メインウィンドウが開いたら、FileメニューからImport Trace File…を選び、BEASTが作成したPrimates.logというファイル(図13)を選択します。
MCMCは確率アルゴリズムなので実際の数値は図に描いたものと全く同じではないことを覚えておいてください。 事後値 (これは樹木尤度と事前密度の積の自然対数) と連続パラメータのトレースがあります。 左側のトレースを選択すると、選択したタブに応じて右側にそのトレースに関する分析結果が表示されます。 最初に開いたときは、「posterior」トレースが選択されており、このトレースの様々な統計量が「Estimates」タブに表示されます。ウィンドウの右上には、選択したトレースの統計量の計算表が表示されます。
左側のリストで clockRate パラメータを選択すると、平均進化速度(全木と全サイトの平均)が表示されます。 Tracer は選択された統計量の (限界事後) ヒストグラムをプロットし、平均や中央値などのサマリー統計量も表示します。 95% HPD は、最高事後密度区間を意味し、選択されたパラメータの事後確率の 95% を含む最もコンパクトな区間を表します。 これは,ベイズ的な信頼区間と大まかに考えることができる. TreeHeightパラメータは、木全体のルートの年齢の周辺事後分布を与えます。
TreeHeightパラメータを選択し、Ctrl-click mrcatime(human-chimp) (Mac OS Xでは Command-click)してください。 すると、ルートの年齢と、先ほどBEAUtiで指定したキャリブレーションMRCAが表示されます。 木(mrcatime(human-chimp))の較正に使ったダイバージェンスが、指定した事前分布と一致する事後分布を持つことが確認できます(図14)。
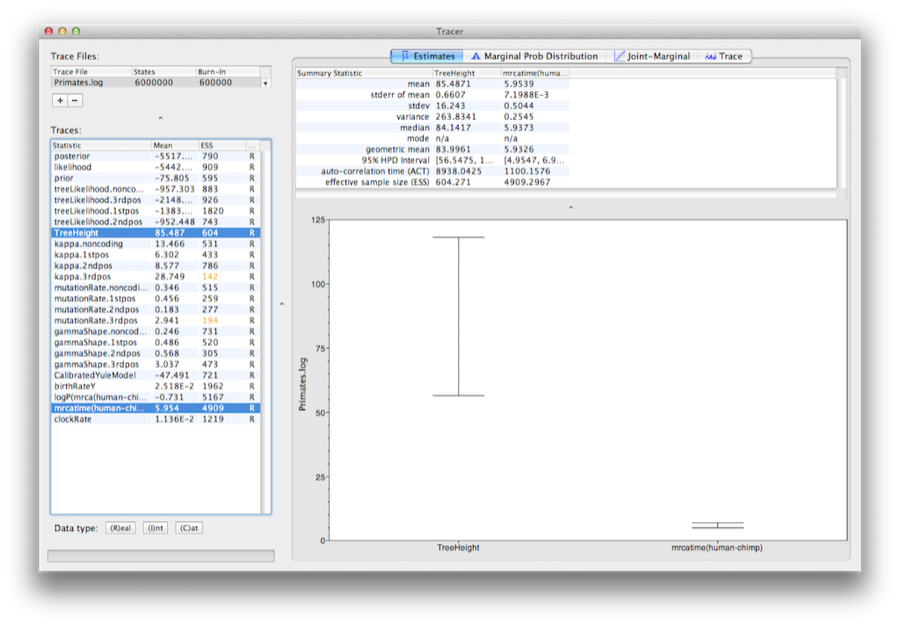
図14:トレーサーでのルートの高さとユーザ指定(human-chimp)MRCAの95%HPD区間を示すスクリーンショットです。
5 Marginal posterior estimates
4分割の相対率を表示するには、4分割のそれぞれについてmutationRateパラメータを選択し、トレーサーの限界密度タブを選択します。図15は相対置換率に対する限界密度を表示したものです。 この図から、コドン位置1と2は大幅に異なる速度(0.456対0.183)で、相対速度が2.941のコドン位置3よりはるかに遅いことがわかります。 ノンコーディングパーティションはコドン位置1と2の中間の速度(0.346)であった。
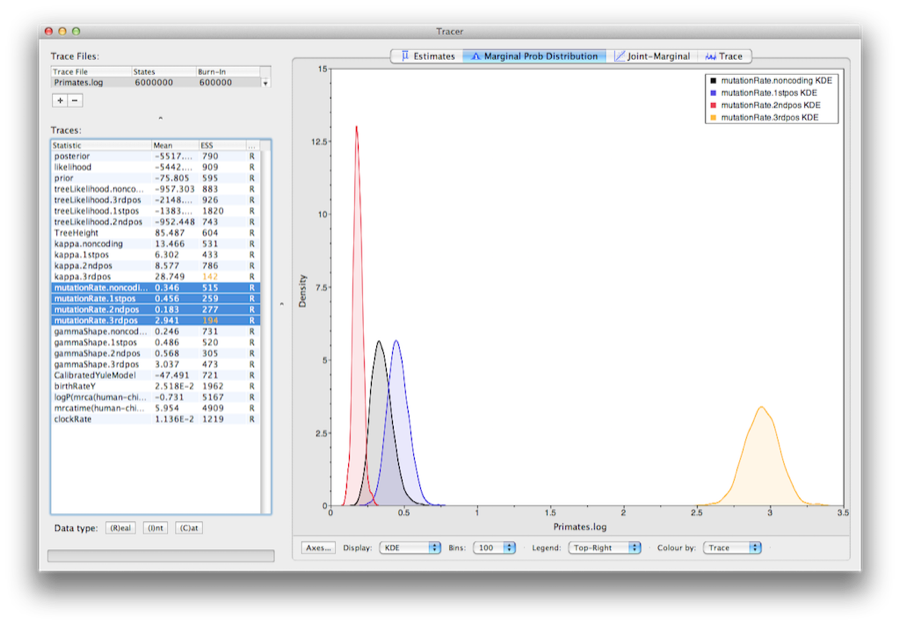
図15:4つのパーティションの相対置換率の限界事後密度(部位加重平均値に対する相対値)のスクリーンショットです。
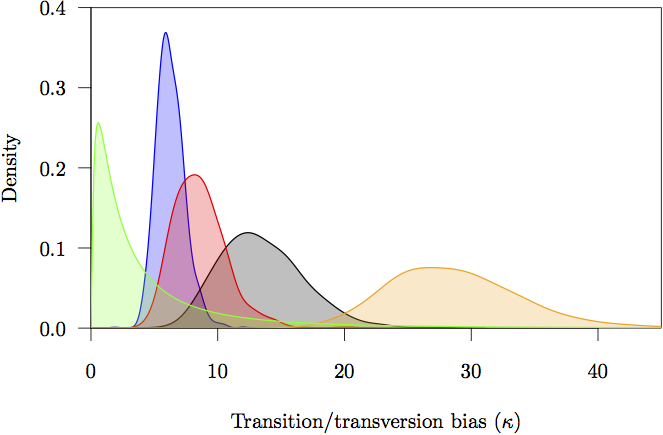
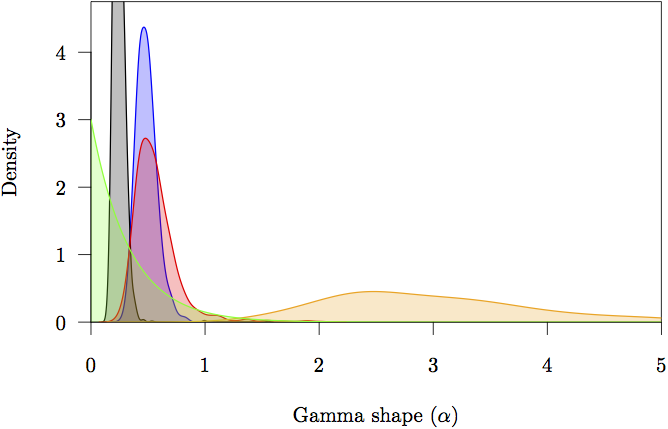
図16:形状(α)パラメータの限界事前密度と事後密度を示したものである。 事前分布はグレーで表示されている。 1134>
図17:遷移・変換バイアス(κ)パラメータの限界事前分布と事後密度。 事前分布はグレーで表示されている。 また、各パーティションの事後密度推定値を示す:非コード(オレンジ)と第1(赤)、第2(緑)、第3(青)のコドン位置。
質問
この遺伝子ツリーの推定分子進化率(95%HPD区間を含む)は何%か?
この推定値にはどのような誤差が含まれていますか。
木のルートは何歳ですか(平均と95%HPDの範囲を与えてください)。
6 系統樹の推定値の取得
BEASTはパラメータ推定値のサンプルとともに系統樹の事後サンプルを作成することも可能です。 これらは、プログラムTreeAnnotatorを使用して要約する必要があります。 これは、木のセットを取り、最も支持されたものを見つけます。 そして、この代表的な樹木に、すべてのthenodeの平均年齢とそれに対応する95%HPDの範囲を注釈する。 また、各ノードの事後的なクレード確率を計算する。 1134>
Figure 18: TreeAnnotatorのスクリーンショット
バーニンはサンプルのスタートから削除する木の本数である。 Tracerがburninとしてsteps数を指定するのとは異なり、TreeAnnotatorでは実際の木の本数を指定する必要がある。 今回の実行では、1,000ステップごとにサンプリングするチェーン長を6,000,000ステップと指定しました。
Posterior probability limit オプションは、ノードがサンプルツリーの中でこの頻度より少なく見つかった場合(つまり、事後確率がこの限界より小さい場合)、そのノードは注釈されないような限界を指定するものです。 デフォルトの0.5は、大多数の木で見られるノードのみが注釈されることを意味する。
Target tree typeは、注釈を付ける木のトポロジーを指定します。 デフォルトのオプションであるMaximum clade credibility treeは、すべてのノードの事後確率の積が最も高いツリーを見つけます。 トポロジーの不確実性が大きく、支持率の低いクレードが多数存在する場合、他の方法では負の枝長を持つ木になる可能性がある。 この分析では、サマリーツリー内のすべてのクレードのサポートが非常に高いので、これは問題ではありません。
入力ファイルには、BEASTが作成した樹木ファイルを選択し、出力用のファイル(ここではPrimates.MCC.treeと呼ぶ)を選択します。
7 推定樹木の視覚化
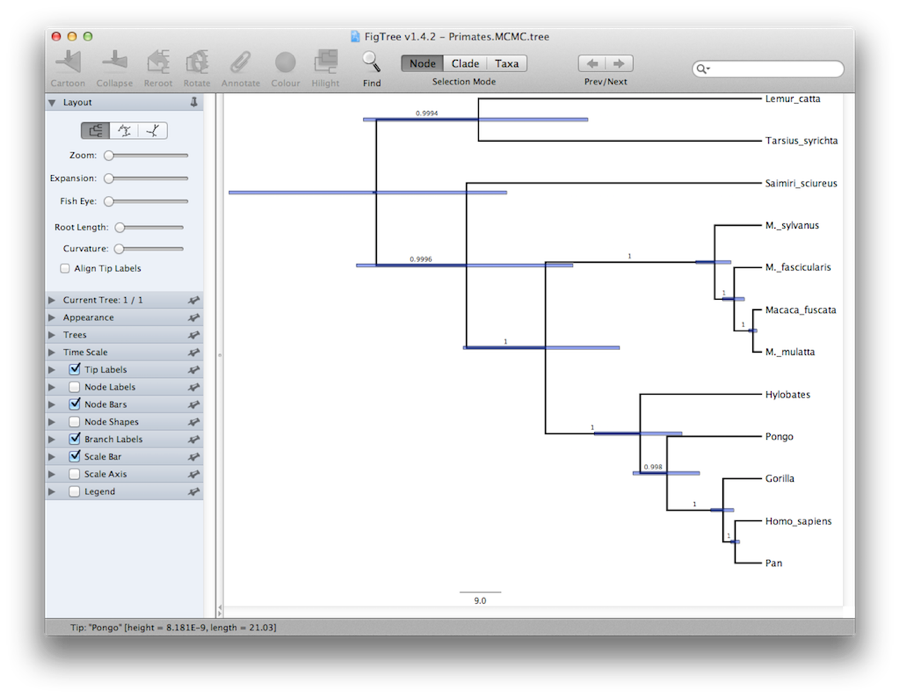
最後に、FigTreeという別のプログラムで樹木を視覚化することができます。 このプログラムを実行し、ファイルメニューの「開く」コマンドでPrimates.MCC.treeファイルを開いてください。 すると、左のコントロールパネルにあるオプションをいくつか選択してみることができます。 まず、パネルにTreesオプションを表示し、Order nodesにチェックを入れ、Ordering by decreasingを選択する。 Node Bars を選択すると、ノード年齢エラーバーが表示されます。 また、Branch Labelsをオンにして、posteriorを選択すると、各ノードの事後確率が表示されます。 1134>
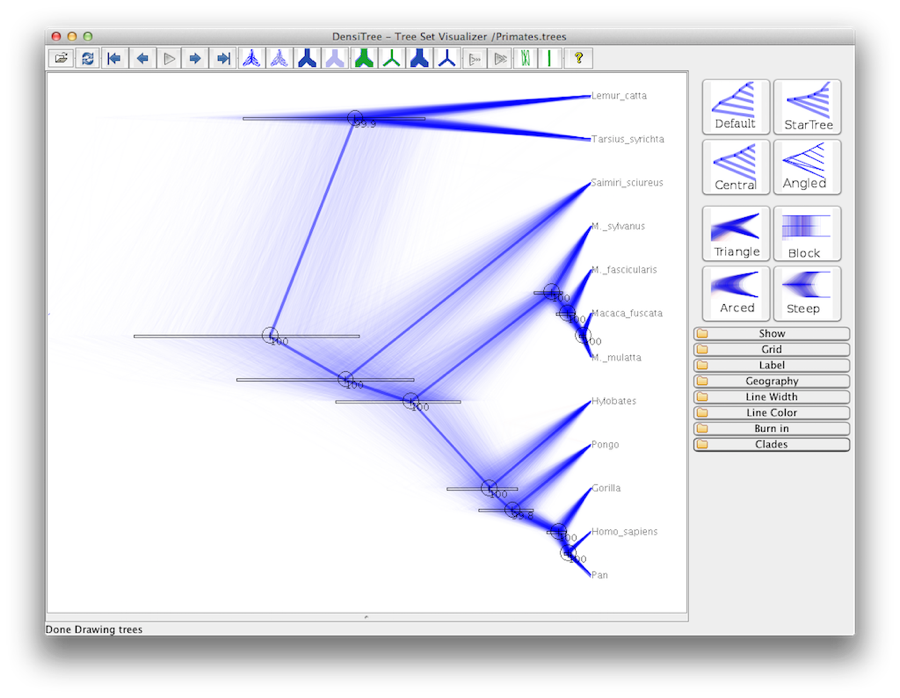
Figure 19: FigTree と DensiTree のスクリーンショットです。
ツリーの別のビューは、Beast 2の一部であるDensiTreeで作成できます。 DensiTreeの利点は、ノードの高さの不確実性とトポロジの不確実性の両方を視覚化できることです。この特定のデータセットでは、優位なトポロジは99%以上のサンプルに存在しています。 そこで、この分析により、トポロジーに関する非常に高いコンセンサスが得られたと結論付けた(図19)。
Questions
- 進化の速度はツリーの異なる系統間で大幅に異なるのか?
- DensiTreeにはクレードバー(メニューウィンドウ/クレードツールバーの表示)があり、クレードの情報を表示します。
クレードの支持率は?
- DensiTreeのトポロジーは「参照」メニューでブラウズすることができます。最も人気のあるトポロジーは99%以上支持されています。
2番目に人気のあるトポロジーの支持率は?
- ヘルプメニューの下に、DensiTreeはいくつかの情報を表示しています。
8 結果を事前分布と比較する
事前分布間の相互作用が事前情報に影響を与えていないことを確認するために、事前分布からサンプリングしながら解析を再実行するのは良いアイデアと言えます。 BEAUtiを使って、同じ解析をセットアップしますが、MCMCオプションのところで、Sample from prior onlyオプションを選択します。 これは、シーケンスデータがない状態で、完全な事前分布を視覚化することができます。 1134>
本章で述べたような「ノード年代測定」を用いた分岐時間推定は、生態学や進化におけるさまざまな疑問に答えるために適用されてきた。 例えば、化石を用いたノード年代測定は、ソテツの種多様性の決定、顕花植物の進化速度の分析、高温・低温砂漠シアノバクテリアの起源を調査するのに用いられた。 このような状況下において、「藍藻類」は、「藍藻類」の中でも、特に、「高温・低温砂漠性シアノバクテリア」のグローバルバイオゲオグラフィーを決定している。 また、このような研究成果をもとに、「ベイズ型ランダム局所時計」、「すべてを支配する一つの速度」、「BMC biology 8 (2010), no.1, 114. A Gelman, G Roberts, and W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little, and S Mathews,Recent synchronous radiation of a living fossil, Science 334(2011), no.6057, 796-799.を発表。 哺乳類ゲノム内およびゲノム間における遷移突然変異の偏りのパターン、Molecularbiology and evolution 20 (2003), no.6, 988-993. このような場合、「哺乳類ゲノムにおける遷移突然変異の偏りのパターン」、「Molecularbiology and evolution 20 (2003), no. 5898, 86-89.
この文書はLATEXからHEVEAによって翻訳されました。