John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, SUA
2Department of Biological Sciences, Kent State University, Ohio, Ohio, SUA
Abstract
Creșterea dovezilor evidențiază relația intimă dintre diabetul de tip II (T2D) și boala Alzheimer (AD). În mod important, aceste două boli au în comun o serie de similitudini patologice, inclusiv acumularea de amiloid, stresul oxidativ, inflamația și moartea celulară. Până în prezent, lipsesc terapiile medicamentoase pentru AD și T2D și există o nevoie crucială de descoperire și dezvoltare de noi terapii pentru aceste boli. O serie de studii la om și la rozătoare au demonstrat că suplimentarea hormonilor metabolici este extrem de valoroasă pentru îmbunătățirea funcției cognitive și a sănătății metabolice generale atât în cazul T2D, cât și în cazul bolii Alzheimer. Amilina, hormonul pancreatic, a apărut ca o componentă crucială a etiologiei bolii, atât a T2D, cât și a DA, deși rolul exact pe care îl joacă amilina în aceste boli nu este încă bine înțeles. Aici, trecem în revistă în mod critic literatura de specialitate actuală care utilizează amilina umană sau analogul său sintetic, pramlintida, precum și antagoniștii receptorilor de amilină pentru tratamentul DA.
Introducere
Boala Alzheimer (DA) este o boală neurodegenerativă progresivă, debilitantă, caracterizată prin acumularea de plăci de amiloid-beta (Aβ) și încurcături neurofibrilare compuse din tau1 hiperfosforilat. Acumularea acestor peptide patologice contribuie la deficite ale funcțiilor executive, cum ar fi învățarea și memoria, starea de spirit, afectivitatea etc. și reprezintă o povară substanțială pentru pacient și îngrijitori. Incidența DA crește într-un ritm alarmant în SUA, estimându-se că 5,5 milioane de americani trăiesc cu DA în 2017 și se așteaptă ca acest număr să se tripleze până în 20502. În plus, costul îngrijirii și tratării pacienților cu DA depășește în prezent 200 de miliarde de dolari anual și se preconizează că va crește3. Deși AD este în mod clar o problemă monumentală în SUA și nu numai, opțiunile de tratament rămân foarte limitate4. Au fost efectuate numeroase studii medicamentoase cu o gamă largă de abordări specifice, însă în prezent există doar șase medicamente aprobate de FDA pentru DA și sunt doar tratamente simptomatice5, 6. Până în prezent, majoritatea agenților farmacologici dezvoltați au vizat în mod specific patologia caracteristică Aβ sau tau, însă niciunul nu a avut succes în eliminarea sau prevenirea patologiei4. Ca atare, există o nevoie fundamentală de a dezvolta tratamente terapeutice și preventive viabile pentru DA.
DDA legată de vârstă (sporadică) este o boală multifactorială complicată, având numeroase influențe genetice și de mediu. Mediul și stilul de viață sunt puternic implicate în dezvoltarea DA sporadică; factori precum dieta7-9, obezitatea8-10, sindromul metabolic7, diabetul de tip II (T2D)9, 11 și bolile cardiovasculare12 au fost toți implicați în cauzalitatea DA. De o importanță crucială, ratele de obezitate și diabet cresc rapid în paralel cu DA12, 13. Deși relația dintre obezitate și DA este oarecum neclară, există dovezi că obezitatea la mijlocul vieții joacă un rol în dezvoltarea DA10. Mai important, obezitatea este frecvent însoțită de o serie de alte boli, inclusiv boli cardiovasculare, hipertensiune, dislipidemie, T2D, accident vascular cerebral etc.14. Incidența T2D este în creștere rapidă, CDC estimând că aproximativ 30,3 milioane de persoane (1 din 10 adulți) din SUA suferă de diabet și un număr impresionant de 84,1 milioane (1 din 3 adulți) suferă de prediabet, dintre care majoritatea nu sunt conștienți de starea lor. Mai mult, din cauza unei scăderi pe scară largă a activității fizice, care este însoțită de o creștere simultană a consumului de alimente și de o dietă deficitară, se propune ca ratele obezității, ale T2D, ale sindromului metabolic și ale bolilor cardiovasculare să crească doar până la un număr estimat de 600 de milioane de cazuri de T2D la nivel mondial până în 203515.
Cercul de dovezi care implică funcția și boala metabolică în procesul de declin cognitiv și de îmbătrânire este substanțial16, 17. De exemplu, aproximativ 70% dintre persoanele diagnosticate cu T2D raportează tulburări cognitive și un număr substanțial de pacienți cu T2D dezvoltă ulterior demență16, 18-21. Persoanele diagnosticate cu T2D timp de cel puțin cinci ani prezintă un risc semnificativ mai mare de a dezvolta DA în comparație cu cele care suferă de T2D de mai puțin de cinci ani17. Împreună, aceste date sugerează că prevalența din ce în ce mai mare a T2D în rândul populației poate contribui la creșterea ratelor de apariție a DA.
T2D se caracterizează inițial prin niveluri ridicate de glucoză în sânge și de insulină, ceea ce duce la hiperinsulinemie; important este faptul că amilina, un mic hormon metabolic produs de celulele β-isletine ale pancreasului, este co-pachetată și cosecretată împreună cu insulina și, prin urmare, este produsă în exces în T2D22. Este important faptul că există o serie de caracteristici patologice care sunt prezente atât în T2D, cât și în AD: 1) scăderea metabolismului cerebral și rezistența hormonală metabolică 2) patologia amiloidă 3) stresul oxidativ (SO) și inflamația. Hiperinsulinemia cronică, hiperinsulinemie și hiperamilinemie duce la o serie de probleme fiziologice: hiperinsulinemia cronică duce la rezistența sistemică la insulină22, la afectarea transportului de insulină prin bariera hemato-encefalică (BBB)23, 24 și, prin urmare, la scăderea semnalizării insulinei în creier25. Pierderea semnalizării insulinei în creier este asociată cu o serie de caracteristici patologice legate de AD, inclusiv producția crescută de Aβ, fosforilarea tau și neuroinflamarea.
În plus, amilina împărtășește caracteristici patologice similare cu Aβ la concentrații ridicate26 și poate fi o cale comună între cele două boli. De exemplu, fibrile de amilină au fost găsite în pancreasul a 95% dintre pacienții cu T2D27-29 și cauzează o serie de perturbări fiziologice, inclusiv influxul aberant de Ca2+, secreția crescută de citokine proinflamatorii30,31 și, în cele din urmă, pierderea celulelor β-izlet32. În plus, amilina traversează cu ușurință BHE și formează fibrile de amilină, precum și plăci mixte cu Aβ în creier, putând fi responsabilă de patologia asemănătoare cu AD și de însămânțarea Aβ în T2D33-35. Se știe că amilina afectează potențarea pe termen lung (LTP) în hipocampus și poate avea o influență înnăscută asupra funcției cognitive din creier36-39. Cu toate acestea, rămâne neclar dacă amilina este o insultă toxică în aceste boli sau dacă pierderea sa funcțională prin agregare sau prin pierderea celulelor β în stadiul tardiv în T2D contribuie la dezvoltarea unei DA.
Dihotomia semnalizării amilinei
Există încă multe dezbateri cu privire la implicarea receptorului amilinei (AMYR) și a semnalizării amilinei în evoluția bolii și etiologia T2D și a DA. Corpul de cercetări care vizează discernerea acestei relații se extinde rapid. Toate cercetările relevante au demonstrat în mod constant că modularea semnalizării amilinei afectează patologia legată de DA. Cu toate acestea, natura acestei relații nu a fost încă elucidată în mod concret. Mai multe grupuri au produs date convingătoare care sugerează că semnalizarea amilinei este benefică în prevenirea patologiei legate de DA și a deficitelor cognitive atât in vivo, cât și in vitro40-44. Este important faptul că pramlintida, o formă recombinantă neagregantă de amilină, utilizată împreună cu terapiile cu insulină pentru tratarea diabetului și care îmbunătățește controlul glicemic, reduce greutatea corporală și reduce markerii serici de OS45-47 , se arată, de asemenea, promițătoare ca terapie pentru DA. Cu toate acestea, până în prezent, nu au existat studii clinice care să vizeze utilizarea amilinei sau a pramlintidei ca agent terapeutic în tratarea demenței. Dovezile clare din studiile pe rozătoare sugerează că tratamentul cronic fie cu amilină umană, fie cu pramlintidă prezintă un puternic beneficiu terapeutic în reducerea patologiei legate de DA; suplimentarea cu amilină/pramlintidă reduce nivelurile de Aβ solubilă, încărcătura plăcii, fosforilarea tau, neuroinflamarea și OS, îmbunătățind în același timp cogniția40-42,44. Datele de mai sus sugerează că o pierdere a semnalizării înnăscute a amilinei în SNC din cauza agregării dă naștere unui risc crescut de dezvoltare a DA și este abordată mai detaliat în Grizzanti et al. 201848.
În schimb, studiile arată, de asemenea, că amilina umană și Aβ au efecte toxice similare și că aceste efecte toxice pot fi atenuate cu ajutorul antagonistului AMYR36-39,49. De exemplu, datele arată că tratamentul in vivo cu antagoniști AMYR produce beneficii fiziologice foarte asemănătoare cu tratamentul cu amilină sau cu pramlintide. Tratamentul șoarecilor TgCRND8 AD cu AC253, un antagonist AMYR, sau cu omologul său ciclic cAC253 reduce neuroinflamarea, nivelurile de Aβ solubilă și încărcătura plăcii, îmbunătățind în același timp cogniția50. În mod similar, studiile in vitro/ex-vivo arată că dozele mici de amilină umană sau Aβ provoacă perturbări ale LTP și că aceste deficite sunt blocate de AC253 sau pramlintide38,39, iar dozele mai mari de amilină umană/oligomeri de amilină sunt asociate cu influxul necontrolat de Ca2+, care este strâns legat de moartea celulară26,32. Împreună, aceste date susțin o funcție toxică a oligomerilor de amilină și, prin urmare, un potențial mecanism terapeutic pentru blocarea AMYR. În schimb, alții au arătat că efectele benefice ale amilinei pot fi blocate cu ajutorul AC25341. Astfel, potențialul terapeutic al tratamentului sau inhibării amilinei rămâne neclar și evidențiază natura complexă și dihotomică a amiloidelor în creier și la periferie.
Piecing Together the Puzzle
Există o serie de lacune în literatura de specialitate actuală care trebuie umplute pentru a oferi o imagine mai completă a poveștii amilinei: 1) natura sistemului înnăscut de amilină și a semnalizării amilinei în creier 2) capacitățile de semnalizare a Aβ și a pramlintidei prin intermediul celor trei receptori AMYR principali și a receptorilor înrudiți 3) mecanismele terapeutice prin care sunt mediate inhibarea amilinei/pramlintidei sau a AMYR. În primul rând, date noi și interesante demonstrează că AMYR nu este implicat doar în semnalizare, ci și în transportul ligandului prin BHE. AMYR este un receptor heterodimeric care este compus dintr-un receptor de calcitonină și o proteină modificatoare a activității receptorului (1-3)51. În acest scop, o reducere globală cu 50% a receptorului de calcitonină (o componentă cheie a AMYR) a redus semnificativ cantitatea de AC253 găsită în creier50, ceea ce indică faptul că AMYR localizat în BBB este implicat în transportul acestor liganzi în creier și poate fi, de asemenea, implicat în deplasarea amilinei și a pramlintidei în/din creier. Existența acestor mecanisme de transport în BBB sugerează că amilina are probabil o funcție fiziologică înnăscută în creier, deoarece transportul său în creier este strâns controlat. Cu toate acestea, modul în care semnalizarea amilinei sau lipsa acesteia duce la caracteristicile patologice ale DA și dacă AMYR este vehiculul prin care Aβ își mediază efectele toxice rămâne încă neclar.
În continuare, există dovezi contradictorii în ceea ce privește relația dintre Aβ și AMYR. Deși mai multe studii demonstrează în mod clar că amilina umană și Aβ au efecte similare asupra LTP în SNC și că utilizarea inhibitorilor AMYR ameliorează aceste efecte dăunătoare36-39, alte dovezi sugerează că Aβ (1-42) este incapabil să semnalizeze prin oricare dintre AMYR pentru a evoca orice fel de răspuns cAMP la o mare varietate de concentrații52. Este posibil ca Aβ să activeze diferite cascade de semnalizare prin interacțiunea cu AMYR sau să acționeze pur și simplu ca un inhibitor competitiv inert, dar acest lucru nu a fost încă demonstrat.
În plus, un studiu separat a demonstrat că amilina oligomerică își mediază efectele toxice direct prin AMYR și indirect prin TRPV4, un canal cationic neselectiv26. Concentrațiile scăzute de amilină umană evocă un răspuns la Ca2+ care este mediat prin intermediul receptorului său nativ. Cu toate acestea, la concentrații mai mari, amilina umană formează oligomeri și activează o semnalizare aberantă care duce la activarea canalelor TRVP4 și permite un aflux necontrolat de cationi, în special de Ca2+. Blocarea farmacologică a AMYR și TRPV4 demonstrează că ambii receptori sunt necesari pentru ca amilina umană oligomerică să inducă efectele sale toxice asupra Ca2+26. Ca atare, este probabil ca Aβ să își medieze efectele toxice asupra AMYR într-un mod similar, deși aceste date nu există încă. Afluxul necontrolat de Ca2+ este legat de o serie de fenomene patologice, inclusiv eliberarea veziculară necontrolată, disfuncția SO și mitocondrială, apoptoza etc. În acest scop, este probabil ca disfuncția celulară și dezvoltarea unei patologii suplimentare asemănătoare cu AD, care rezultă din semnalizarea toxică a amiloidului, să fie mediate atât prin AMYR, cât și prin TRPV4. Ca atare, este necesar să se distingă cascadele de semnalizare care modulează relația dintre AMYR și TRVP4. În plus, sunt justificate experimentele farmacologice care utilizează Aβ și pramlintida pe o gamă largă de doze pentru a determina efectele Aβ și pramlintidei asupra curenților de Ca2+, LTP, producția de cAMP și alte cascade de semnalizare pentru a determina capacitățile lor de semnalizare. Aceste experimente vor contribui la umplerea unora dintre lacunele din literatura de specialitate actuală în ceea ce privește AMYR și implicarea sa în stările de boală (figura 1).
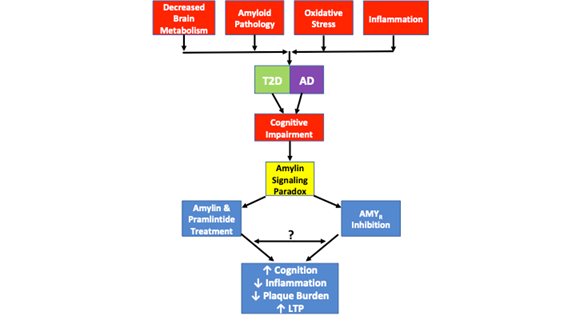
Figura 1. Reprezintă paradoxul semnalizării amilinei și similitudinile patologice observate în T2D și AD. Scăderea metabolismului cerebral, patologia amiloidă, stresul oxidativ și inflamația sunt toate caracteristici patologice comune observate în ambele boli. Deși nu fiecare caz de T2D sau AD include fiecare dintre aceste caracteristici patologice, fiecare caz prezintă tulburări cognitive. Paradoxul semnalizării amilinei intră în joc, deoarece studiile au arătat că atât inhibarea AMYR, cât și agonismul AMYR prin tratamentul cu amilină și pramlintidă au ca rezultat îmbunătățirea cogniției, scăderea inflamației, scăderea încărcăturii plăcilor și creșterea LTP. Mecanismele de semnalizare guvernate de agonismul AMYR și antagonismul AMYR nu au fost încă pe deplin elucidate. În timp ce semnalizarea amilinei este asociată în mod tradițional cu semnalizarea cAMP și PKA, nu este clar dacă alte cascade sunt, de asemenea, activate de amilină/pramlintidă. Mai mult decât atât, nu este clar dacă antagoniștii AMYR, oligomerii de amilină sau Aβ semnalizează prin AMYR sau dacă există similitudini sau diafonie între toți acești liganzi AMYR. Ca atare, o serie de experimente propuse în această recenzie vor contribui la elucidarea în continuare a adevăratei naturi a AMYR.
Concluzii
Disparitatea actuală în ceea ce privește rolul semnalizării amilinei în creier demonstrează o nevoie esențială de elucidare în continuare a implicării amilinei atât în AD cât și în T2D. În cazul T2D, este probabil ca în stadiile incipiente ale bolii, amilina să inunde creierul, să formeze oligomeri, să inducă o semnalizare aberantă prin receptorul său nativ și să recruteze TRPV4 pentru a induce influxul patologic de Ca2+ care are ca rezultat o disfuncție neuronală generalizată care se manifestă prin OS, eliberare veziculară necontrolată și disfuncție interneuronală, inflamație și moarte celulară. Acest mecanism poate fi responsabil pentru tranziția inițială de la creierul sănătos la îmbătrânirea creierului în cazul bolilor metabolice. Ca atare, inhibarea AMYR sau TRVP4 în anumite momente ale bolii metabolice și în stadiile incipiente ale diabetului ar putea fi justificată pentru a bloca efectele toxice ale amilinei oligomerice sau ale Aβ. Cu toate acestea, dovezi solide sugerează, de asemenea, că înlocuirea amilinei, fie cu amilină umană, fie cu pramlintidă, reduce majoritatea patologiilor majore legate de DA, îmbunătățind în același timp cogniția în modelele de DA la rozătoare. Ca atare, ar putea fi justificată înlocuirea semnalizării amilinei cu amilină sau pramlintide în stadiile medii și târzii ale diabetului, atunci când semnalizarea amilinei este pierdută din cauza agregării, oligomerizării sau pierderii celulelor β. În acest scop, este necesar, de asemenea, să se discearnă prezentarea temporală a evenimentelor patologice în cazul îmbătrânirii creierului legate de metabolism și opțiunile terapeutice pentru stadiile timpurii, intermediare și târzii ale bolii. Analiza și testarea critică a naturii directe și a capacităților de semnalizare ale acestor amiloizi, precum și natura terapeutică a tratamentelor temporale specifice pot contribui la reducerea decalajului dintre terapiile de inhibare a AMYR și terapiile de înlocuire a amilinei.
Finanțare
Finanțarea pentru acest articol a fost asigurată de National Institutes of Aging grant 1R15AG050292-01A1.
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau și disfuncția sinaptică. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurologie. 2013; 80: 1778-1783.
- Asociația As. 2016 Alzheimer’s disease facts and figures. Alzheimer’s & Demența. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: puțini candidați, eșecuri frecvente. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: systematic review and economic model. Îmbătrânirea vârstei. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glucose levels and risk of dementia. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of the dementia prevalence in the United States and China. Obesitate. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Jurnalul de investigare a diabetului. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Diabetul de tip 2 ca factor de risc pentru boala Alzheimer: factorii de confuzie, interacțiunile și neuropatologia asociată cu această relație. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurologie. 2011; 77: 461-468.
- Ginter E, Simko V. Prevalența globală și viitorul diabetului zaharat În Diabetes Springer. 2013; 35-41.
- Federation ID. Atlasul de diabet al IDF. Bruxelles: Federația Internațională de Diabet. 2013.
- K Dash S. Tulburarea cognitivă și diabetul. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Riscul de demență în rândul persoanelor cu diabet zaharat: un studiu de cohortă bazat pe populație. Am J Epidemiol. 1997; 145: 301-308.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalența diabetului și a prediabetului și a factorilor lor de risc în rândul adulților din Bangladesh: un studiu la nivel național. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetul zaharat și riscul de demență The Rotterdam Study. Neurologie. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Enzima de degradare a insulinei reglează nivelurile de insulină, proteina β-amiloidă și domeniul intracelular al proteinei precursoare β-amiloide in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Legarea insulinei la capilarele cerebrale este redusă la șobolanii Zucker obezi genetic, hiperinsulinemici Zucker. Peptide. 1990; 11: 467-472.
- Gil-Bea FJ, Solas M, Solomon A, et al. Nivelurile de insulină sunt scăzute în lichidul cefalorahidian al femeilor cu boala Alzheimer prodomală. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neurofarmacologie. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mecanismele care leagă obezitatea de rezistența la insulină și diabetul de tip 2. Nature. 2006; 444: 840-846.
- Johnson K, O’Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. Jurnalul american de patologie. 1989; 135: 245.
- Johnson KH, O’Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. β-amiloidul Alzheimer, amilina din insulele umane și fragmentul proteinei prion evocă creșteri ale calciului liber intracelular printr-un mecanism comun într-o linie celulară neuronală GnRH hipotalamică. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Depunerea de amilină în creier: un al doilea amiloid în boala Alzheimer. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. Însămânțarea in vivo și însămânțarea încrucișată a amiloidozei localizate: o legătură moleculară între diabetul de tip 2 și boala Alzheimer. Jurnalul american de patologie. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Receptorul amilinei: o țintă fiziopatologică comună în boala Alzheimer și diabetul zaharat. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ) peptide activează direct subtipul de receptor amilin-3 prin declanșarea mai multor căi de semnalizare intracelulară. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide Antagonizează depresia indusă de beta-amiloid (Aβ) și amilina umană a potențierii pe termen lung a hipocampului indusă de amilina umană. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Depresia indusă de beta-amiloid a potențării pe termen lung a hipocampului este mediată prin intermediul receptorului amilinei. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Injecția intraperitoneală a peptidei pancreatice amilină reduce puternic afectarea comportamentală și patologia amiloidă a creierului în modelele murine de boală Alzheimer. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Liganzii receptorilor de amilină reduc cascada patologică a bolii Alzheimer. Neurofarmacologie. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Efectele neuroprotectoare ale analogului amilinei pramlintide asupra patogenezei bolii Alzheimer și a cogniției. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilizarea β-cateninei prin mutații în presenilina-1 potențează apoptoza neuronală. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Tratamentul cu amilină reduce neuroinflamarea și ameliorează modelele anormale de expresie a genelor în cortexul cerebral al unui model de șoarece cu boala Alzheimer. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetat injectabil pentru tratamentul diabetului zaharat de tip 1 și de tip 2. Clin Ther. 2007; 29: 535-562.
- Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta?analysis. Diabetes, Obesity and Metabolism (Diabet, obezitate și metabolism). 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Efecte neuroprotectoare ale analogilor de amilină asupra patogenezei și cogniției bolii Alzheimer. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Acțiunile proteinei β-amiloide asupra neuronilor umani sunt exprimate prin intermediul receptorului de amilină. Jurnalul american de patologie. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyclic AC253, un nou antagonist al receptorului amilinei, îmbunătățește deficitele cognitive într-un model de șoarece al bolii Alzheimer. Alzheimer’s & Demența: Translational Research & Intervenții clinice. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reducerea comportamentului nociceptiv la șoarecii knockout ai polipeptidei amiloide a insulelor (amilină). Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Activitatea pramlintidei, a amilinei umane și de șobolan, dar nu și a Aβ1-42 la receptorii amilinei umane. Endocrinologie. 2014; 155: 21-26.
Khaodhiar L, McCowen KC, Blackburn GL. Obezitatea și afecțiunile sale comorbide. Clin Cornerstone. 1999; 2: 17-31.
Biessels GJ, Strachan MW, Visseren FL, et al. Demența și declinul cognitiv în diabetul de tip 2 și în stadiile prediabetice: către intervenții țintite. The lancet Diabetes & Endocrinologie. 2014; 2: 246-255.
Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
Masters SL, Dunne A, Subramanian SL, et al. Activarea inflammasomului NLRP3 de către polipeptidul amiloid al insulelor oferă un mecanism pentru creșterea IL-1β în diabetul de tip 2. Nat Immunol. 2010; 11: 897.