Introducere
Mutațiile de novo (DNM) din linia germinală sunt modificări genetice ale individului cauzate de mutageneza care are loc în gameții parentali în timpul oogenezei și spermatogenezei. Aici, termenul „de novo” nu trebuie confundat cu cel de „mutație nouă”. În ciuda faptului că DNM-urile în contextul unui trio (tată, mamă și copil) sunt mutații noi, acestea pot fi variante comune, rare sau noi în populația generală. Pentru a măsura și a explica rata unei anumite DNM, este necesar să se evalueze mai întâi impactul asupra fenotipului variantei, deoarece noi trăsături favorabile pot evolua atunci când mutațiile genetice apărute oferă un beneficiu specific de supraviețuire (Front Line Genomics, 2017).
La oamenii cu boli genetice nemendeliene care apar sporadic, DNM-urile sunt de obicei noi, mai fiabile și mai dăunătoare decât variantele moștenite, deoarece nu sunt supuse unei selecții naturale puternice (Crow, 2000; Front Line Genomics, 2017). Prin urmare, identificarea cauzei genetice a unei tulburări induse de un DNM la un individ poate fi o provocare din punct de vedere clinic, deoarece pleiotropia și eterogenitatea genetică pot sta la baza unui singur fenotip (Eyre-Walker și Keightley, 2007). În consecință, în ultimul deceniu, s-au depus eforturi considerabile pentru a secvenția exomii de la indivizi cu boli cu etiologie genetică neclară în scopul diagnosticării clinice. Cu toate acestea, chiar și după detectarea variantelor de novo candidate, există încă informații insuficiente cu privire la variantele comune și rare, ceea ce împiedică o concluzie clară cu privire la patogenitatea variantei de novo identificate și la rolul acesteia în boală (Acuna-Hidalgo et al., 2016). Această limitare poate fi explicată prin faptul că variantele de novo sunt, de obicei, heterozigote și pot fi fie extrem de rare, fie comune. În cazul variantelor de novo foarte rare, patogenitatea variantei poate fi greu de dovedit, deoarece nu mai există alți pacienți cu același fenotip și variantă de novo. În cazul variantelor de novo comune, este posibil să nu se cunoască factorii care determină manifestările de patogenitate ale variantei, în special dacă unii indivizi din populația generală au varianta, dar nu au boala genetică. Cu toate acestea, indiferent de rata variantelor de novo, ambele tipuri de variante pot fi scalate pe baza adecvării relative și a selecției naturale.
Adaptarea depinde de mulți factori; prin urmare, pentru a evalua dacă un DNM este patogen sau adaptativ și pentru a înțelege de ce apare cu o anumită frecvență în populație, este necesar să se examineze varianta în condiții adecvate. Acestea includ mediul, vârsta părinților, contextul genomic, epigenetica și alți factori, deoarece toate acestea influențează valoarea adecvării relative medii care crește monoton, în timp ce intensitatea selecției scade (Peck și Waxman, 2018).
Obiectivul principal al acestui studiu a fost de a elucida rata de apariție a DNM-urilor și de a determina modul în care aceste mutații sunt distribuite în exomii populației generale lituaniene. De asemenea, am examinat dacă frecvența acestor mutații a fost afectată de compoziția sau de parametrii structurali ai secvențelor în care au apărut și de alți factori care ar putea influența mecanismele care stau la baza formării acestor DNM-uri. În cele din urmă, am încercat să stabilim dacă DNM-urile au apărut din cauza presiunii intensive a selecției naturale asupra regiunilor funcționale. Deși distribuția și intensitatea DNM-urilor au fost subiecte ale multor studii, acestea nu fuseseră explorate anterior în populația lituaniană.
Materiale și metode
În acest studiu, am analizat eșantioane din populația lituaniană obținute în cadrul proiectului LITGEN (LITGEN, 2011). Setul de date a fost alcătuit din 49 de trio-uri cu un total de 144 de indivizi diferiți. ADN-ul genomic a fost extras din sângele venos utilizând fie metoda de extracție cu fenol-cloroform, fie platforma automată de extracție a ADN-ului TECAN Freedom EVO® (Tecan Schweiz AG, Elveția) bazată pe metoda particulelor paramagnetice. Exomii au fost secvențiați pe un sistem de secvențiere SOLiD 5500 (citiri de 75 bp). Datele de secvențiere au fost procesate și pregătite de software-ul Lifescope. Exomii au fost cartografiați în conformitate cu genomul uman de referință build 19. O adâncime medie de citire a secvențierii a fost de 38,5. Fișierele în format BAM ale mamei, tatălui și copilului generate de Lifescope au fost combinate cu ajutorul software-ului SAMtools pentru fiecare trio.
Mutațiile de novo au fost identificate de două programe software: VarScan (Koboldt et al., 2012) și VarSeqTM. O variantă potențială a fost considerată a fi un DNM dacă a fost identificată în descendenți, dar nu era prezentă la niciunul dintre părinți în aceeași poziție. În total, 1 752 și 4 756 de DNM au fost detectate de VarScan și, respectiv, VarSeqTM. Pentru a elimina apelurile de novo fals-pozitive, atunci când nu se știa dacă toți indivizii din trio au fost identificați corect, au fost aplicate filtre conservatoare asupra parametrilor de calitate DNM detectați, după cum urmează: (1) calitatea genotipică a individului ≥50; (2) numărul de citiri la fiecare sit >20. Software-ul SnpSift a fost utilizat pentru a aplica aceste filtre pe datele generate de VarScan. Datele generate de software-ul VarSeqTM au fost filtrate prin alegerea acelorași parametri de filtrare în segmentul Trio Workflow. În plus, pentru a elimina variantele rămase care erau somatice (prezente doar într-o fracțiune din celulele sanguine secvențiate), cu un echilibru alelelor scăzut sau artefacte de secvențiere, DNM-urile au fost filtrate prin stabilirea unui prag pentru fracțiunea observată de citiri la indivizii cu alela alternativă (echilibrul alelelor) pentru trio (Kong et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015). În plus, toate variantele posibile identificate și filtrate de novo de un singur nucleotid au fost revizuite manual cu Integrative Genomics Viewer (Robinson et al., 2011). Datorită numărului mare de DNM-uri identificate, pentru validarea variantelor prin secvențiere Sanger, au fost selectate aleatoriu 51 de variante single nucleotide de novo. Secvențierea Sanger a fost efectuată cu ajutorul unui analizor genetic ABI PRISM 3130xl. Toate DNM-urile filtrate și revizuite manual identificate de VarScan (N = 95) și de VarSeqTM (N = 84) au fost adnotate cu ajutorul ANNOVAR (Butkiewicz și Bush, 2016; Wang et al., 2010). Pentru analiza interacțiunilor proteinelor, a fost utilizat software-ul STRING (Szklarczyk et al., 2017). Ca și în cazul cartografierii exomului, adnotările au fost efectuate utilizând genomul uman de referință hg19.
Probabilitatea ca o poziție de apelare să fie un DNM în trio a fost calculată independent pentru fiecare trio. Așa cum a fost descris într-o referință anterioară (Besenbacher et al., 2015), rata de novo pe poziție pe generație (PPPG) a fost calculată după cum urmează:
unde f este numărul de trio-uri și N este numărul de situri apelabile, care pot fi potențial identificate ca situri de novo pentru fiecare trio în parte, indiferent de adâncimea de secvențiere. Acest număr variază în funcție de trio. ni este numărul de DNM-uri identificate pentru trio i. Probabilitatea Pji (nucleotid de novo ingle) pentru ca situl de nucleotid unic apelat j și familia i să fie mutant a fost calculată după cum urmează:
Probabilitatea Pji (de novo indel)pentru ca situsul indel numit j și familia i să fie mutat a fost calculată după cum urmează:
unde C, M și F reprezintă descendența, mama și, respectiv, tatăl, iar Hetero, HomR și HomA semnifică heterozigot, homozigot pentru referință și, respectiv, homozigot pentru alela alternativă. Probabilitatea Pij (de novo) a fost calculată în raport cu acoperirea de secvențiere. Intervalele de încredere pentru estimările ratei au fost calculate ca pentru proporțiile binomiale. Pentru estimarea ratei DNM și pentru calculele ulterioare, am utilizat pachetul R (versiunea 3.4.3) (R Core Team, 2013).
Pentru a testa ipoteza conform căreia variațiile ratei DNM în diferite regiuni ale genomului ar putea fi explicate de caracteristicile intrinseci ale regiunii genomice în sine și de vârsta părinților, s-a efectuat o analiză de regresie liniară, pentru care s-a efectuat adnotarea „secundară” a fiecărui DNM, utilizând date din proiectele ENCODE (ENCODE Project Consortium, 2012) și LITGEN (LITGEN, 2011). În primul rând, în conformitate cu un studiu anterior (Besenbacher et al., 2015), pentru a colecta înregistrări privind peisajul genomic al DNM-urilor identificate, au fost alese liniile celulare limfoblastoide (LCL și GM12878) (ENCODE Project Consortium, 2012). Au fost colectate date pentru:
(1) rate de expresie (eQTL) (ENCODE Project Consortium, 2012; Lappalainen et al., 2013; GTEx Consortium et al., 2017) în diferite țesuturi. În funcție de expresia regiunilor cu DNM-uri au fost împărțite în poziții cu expresie specifică și nespecifică;
(2) măsurători ale situsurilor de hipersensibilitate la DNază1 (DHS). Statutul DHS a fost atribuit 0 dacă se afla în afara vârfului DHS și 1 dacă se afla în interiorul acestuia;
(3) măsurători ale contextului insulelor CpG. Dacă DNM era în interiorul insulelor CpG s-a atribuit un statut de poziție 1; dacă era în afara – 0;
(4) trei mărci histonice (H3K27ac, H3K4me1 și H3K4me3) din proiectul ENCODE. Dacă DNM se afla în poziția marcată cu histone, a fost atribuit cu 1, iar dacă nu – 0;
(5) Valorile de conservare GERPP++ au fost colectate cu ajutorul instrumentului de adnotare ANNOVAR. În funcție de valorile de conservare, pozițiile cu DNM-uri au fost atribuite în poziții conservatoare (scor GERP++ >12) și poziții neconservatoare (scor GERP++ <12) (Davydov et al., 2010; ENCODE Project Consortium, 2012). Pe baza înregistrărilor chestionarului din proiectul LITGEN, au fost colectate date privind vârsta părinților. După colectarea parametrilor pentru fiecare trio, a fost calculat un număr de poziții cu fiecare parametru. Apoi a fost efectuată o analiză de corelație urmată de modelarea prin regresie liniară a ratei DNM și a parametrilor.
Rezultate
După analiza DNM, a fost identificat un număr excepțional de mare de DNM-uri pentru două trio-uri (nr. 4 și 21): 113 și 123 (prin VarScan și VarSeqTM, respectiv) și 16 (VarScan). Aceste constatări ne-au determinat să testăm paternitatea biologică, care a fost respinsă pentru trioul nr. 4 și confirmată pentru trio nr. 21. Astfel, datele pentru trio nr. 4 au fost excluse din studiu. În setul final de 48 de trio-uri, 95 de DNM-uri au fost identificate în 34 de trio-uri cu software-ul VarScan și 84 de DNM-uri în 31 de trio-uri au fost identificate cu software-ul VarSeqTM (figura 1). Nu au fost detectate DNM-uri în 18 și 15 trio-uri cu VarScan și, respectiv, VarSeqTM. Dintre toate DNM-urile identificate de ambele programe software, doar 5,37 % dintre DNM-uri au corespuns (trei DNM-uri în genele MEIS2, PGK1 și MT1B). Fiecare persoană a avut în medie 1,9 (software VarScan) și 1,7 (VarSeqTM) DNM-uri.
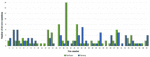
FIGURA 1. Comparația variantelor de novo de un singur nucleotid identificate de software-ul VarScan (albastru) și VarSeqTM (verde).
Analiza celor 95 de DNM-uri care au fost identificate de software-ul VarScan a arătat că 20 de DNM-uri erau exonice, inclusiv două DNM-uri stop-gain, șapte DNM-uri sinonime și 11 DNM-uri nesinonime. Optzeci de mutații noi identificate de VarSeqTM au fost exonice, inclusiv 1 DNM stop-gain și 78 de DNM nesinonime (figura 2). Majoritatea DNM-urilor identificate de VarScan au fost în cromozomii 1, 2, 4 și 5, în timp ce VarSeqTM a identificat DNM-uri predominant în cromozomii 2, 6, 7 și 11. Numărul de DNM identificate nu a fost corelat cu densitatea genelor în cromozomi (R = 0,09, valoare p = 0,65 pentru VarScan și R = 6,73, valoare p = 0,51 pentru VarSeqTM) sau cu dimensiunea cromozomului (figura 3). Conform ambelor programe software, ratele de tranziții și transversiuni au fost foarte asemănătoare: 1,44 și, respectiv, 1,47 (figura 4). Cu toate acestea, au fost identificate diferențe în structurile tranzițiilor. Mai exact, printre DNM-urile identificate de VarScan, au existat mai multe modificări G/T și A/C, în timp ce printre DNM-urile identificate de VarSeqTM, au existat mai multe modificări A/T și G/C.
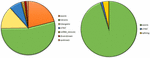
FIGURA 2. Compoziția mutațiilor de novo (DNM) generate de VarScan (în stânga) și de VarSeqTM (în dreapta).
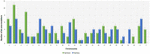
FIGURA 3. Distribuția numărului de variante de novo pe cromozom în funcție de datele generate de VarScan și VarSeqTM. Barele verzi reprezintă DNM-urile identificate de software-ul VarScan, iar cele albastre – de VarSeqTM. Barele de eroare reprezintă eroarea standard a mediei DNM-urilor pentru fiecare cromozom.
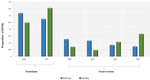
FIGURA 4. Evenimentele moleculare care stau la baza tranzițiilor apar mai frecvent decât cele care conduc la transversiuni, rezultând o rată de ∼1,5 ori mai mare de tranziții față de transversiuni în întregul exom. Evenimente de tranziție și de transversiune identificate de software-ul VarScan (verde) și VarSeqTM (albastru). Barele de eroare reprezintă eroarea standard a mediei DNM-urilor.
Ratele calculate ale mutațiilor de novo de nucleotide unice au fost de 2,4 × 10-8 PPPG (interval de încredere de 95% : 1,96 × 10-8-2,99 × 10-8) conform VarSeqTM și 2.74 × 10-8 per nucleotid pe generație (95% IC: 2,24 × 10-8-3,35 × 10-8) conform VarScan.
Trei indeli de novo în trei triouri au fost identificate de algoritmul VarScan în cromozomii 6 și 11. Rata calculată a indelurilor de novo în genom a fost de 1,77 × 10-8 (95% CI: 6,03 × 10-9-5,2 × 10-8) PPPG. În mod notabil, toate indelurile de novo au fost „reversibile”, adică părinții au avut noi variante în genom, iar copiii lor au avut variante de novo pe baza genomului de referință cu valoarea medie de 37,5 a adâncimii de secvențiere și, respectiv, 50 de calitate a genotipurilor. Cu toate acestea, aceste trei DNM-uri nu au fost selectate pentru validarea prin metoda de secvențiere Sanger, astfel rămâne totuși o probabilitate de supraestimare a indelurilor de novo. Indelurile de novo au fost C/T și A/G în contextul unor singure nucleotide.
Modelarea regresiei liniare a arătat că situsurile de hipersensibilitate DNAse 1, contextul insulelor CpG, valorile de conservare GERPP++ și nivelurile de expresie au explicat ∼68-93% din ratele DNM (tabelul 1). Nici markerii epigenetici și nici vârsta paternă nu s-au corelat semnificativ cu rata DNM. Modelele au fost stabilite doar pe baza datelor obținute din VarScan, deoarece nu a existat nicio corelație între datele din VarSeqTM și caracteristicile intrinseci ale regiunii genomice în sine.

TABEL 1. Regresia liniară a situsurilor de hipersensibilitate DNAaseI, a contextului insulelor CpG, a valorilor de conservare GERPP++ și a efectului nivelului de expresie al asupra ratei DNM-urilor.
Predicția funcțională a DNM-urilor
Pentru a evalua care mutații missense au fost dăunătoare și au alterat funcția proteinei afectate în funcție de tip, au fost analizate scorurile categoriale prezise pentru daunele induse de DNM-uri. Au fost luate în considerare următoarele 10 valori: polyphen HDIV și HVAR, LRT, PROVEAN, CADD, FATHMM, Mutation Taster, MutationAssessor, SIFT, codarea Fathmm-MKL și GERP++. Pe baza scorurilor prezise, au fost selectate patru DNM identificate de VarScan ca având șase sau mai multe predicții dăunătoare sau probabil dăunătoare. Aceste DNM-uri stop-gain se aflau în genele MEIS2 și ULK4, în timp ce DNM-urile nesinonime se aflau în genele MT1B și PGK1. Proteinele codificate de aceste gene sunt importante pentru creșterea neuronală, endocitoza și protecția împotriva efectelor negative ale metalelor grele. Aceste proteine participă la eliberarea angiostatinei, inhibitor al vaselor sanguine tumorale, și la diverse căi de semnalizare. Nu au existat conexiuni între proteinele codificate de aceste gene (figura 5).
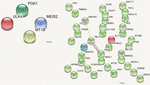
FIGURA 5. Interacțiuni proteină-proteină (Szklarczyk et al., 2017) în genele care adăpostesc DNM-uri. DNM-urile identificate prin VarScan în genele care codifică proteine sunt în stânga, DNM-urile identificate prin VarSeqTM – în dreapta. Liniile colorate indică o legătură între proteine.
Mutațiile de novo identificate prin VarSeqTM au fost analizate mai în detaliu dacă au fost prezise ca fiind dăunătoare sau probabil dăunătoare de cel puțin jumătate dintre instrumentele de predicție. Au existat 35 de mutații punctiforme (a se vedea ??) în gene care codifică proteine importante pentru remodelarea cromatinei, reglarea citoscheletului, creșterea și viabilitatea celulelor, căile de semnalizare citoplasmatică și inițierea răspunsurilor neuronale care declanșează percepția mirosului.
Printre proteinele codificate de genele afectate de DNM, doar CLPTM1, ZNF547 și DMXL1 au fost conectate într-un anumit fel (Figura 5).
Discuție
În acest studiu, am efectuat o analiză cuprinzătoare a distribuției DNM-urilor în diferite regiuni ale exomului în populația lituaniană. În total, au fost detectate 95 DNM-uri în 34 de trio-uri și 84 DNM-uri în 31 de trio-uri cu ajutorul tehnologiei de secvențiere SOLiD 5500 prin intermediul algoritmilor VarScan și, respectiv, VarSeqTM. În primul rând, am dori să remarcăm faptul că am ales VarScan pentru apelarea DNM-urilor deoarece, conform (Warden et al., 2014), acest algoritm produce o listă de variante, cu o concordanță ridicată (>97%) cu variantele de înaltă calitate apelate de GATK UnifiedGenotyper și HaplotypeCaller. Software-ul VarSeqTM a fost ales deoarece este un instrument utilizat pe scară largă pentru analiza variantelor, atât în cercetări, cât și în analize clinice. În ciuda faptului că ambii algoritmi au fost concepuți pentru a căuta DNM-uri în exomul descendenților care nu erau prezente la niciunul dintre părinți, acordul dintre cele două programe software pentru analiza DNM-urilor a fost de numai 5,37%. Algoritmul VarScan a avut o sensibilitate mai mare (5,42%) pentru detectarea DNM înainte de filtrare decât algoritmul VarSeqTM (1,77%), astfel, am suspectat că niciun instrument nu a reușit să cheme mutații din cauza sensibilității ridicate, care a fost întotdeauna însoțită de o specificitate scăzută. Prin urmare, sugerăm că o îmbunătățire considerabilă a rezultatelor ar putea fi obținută prin combinarea rezultatelor diferitelor instrumente (Sandmann et al., 2017).
Pe baza datelor generate, rata estimată a DNM de un singur nucleotid a fost între 2,4 × 10-8 și 2,74 × 10-8, iar cea a indelurilor de novo a fost de 1,77 × 10-8 PPPG, în funcție de algoritmul utilizat. Rata DNM calculată de noi a fost mai mare decât cea raportată în studiile anterioare (Kong et al., 2010, 2012; Neale et al., 2012; Szamecz et al., 2014; Besenbacher et al., 2015; Francioli et al., 2015), în care a variat între 1,2 × 10-8 și 1,5 × 10-8 PPPG. Rata DNM mai mare din studiul nostru a fost rezonabilă, deoarece studiul nostru s-a bazat pe date exome. În plus, exomii prezintă rate de mutație semnificativ mai mari (cu 30 %) decât genomurile întregi, deoarece compoziția perechilor de baze a genomului întreg este diferită de cea a exomilor. În special, exomii au un conținut mediu de GC de aproximativ 50 %, în timp ce cel al întregului genom este de aproximativ 40 % (Neale et al., 2012). CpG-urile metilate reprezintă secvențe extrem de mutabile la om datorită dezaminării spontane a bazelor de citosină (Neale et al., 2012). Conform studiilor de genomică comparativă, se crede că ratele crescute de mutație în regiunile bogate în CpG au evoluat în jurul perioadei de iradiere a mamiferelor (Francioli et al., 2015). În timpul divergenței speciilor, regiunile exonice bogate în CpG au suferit rate de mutație crescute în comparație cu cele de la nivelul ADN-ului necodificator și s-au transformat în regiuni necodificatoare. Prin urmare, apoi efectul conținutului de CpG scade în timp, rata medie de mutație scade până când ajunge la nivelul prezent în ADN-ul necodificator din jur (Subramanian și Kumar, 2003). Cu toate acestea, în timp ce secvențele din regiunile cu evoluție neutră ale genomului au avut suficient timp să se echilibreze în ceea ce privește contextele dinucleotide, selecția purificatoare a menținut CpG-uri hipermutabile în regiunile funcționale (Subramanian și Kumar, 2003; Schmidt et al., 2008; Francioli et al., 2015). Prin urmare, deoarece am constatat o rată DNM mai mare decât cea raportată de alte studii, am speculat că aceasta s-ar putea datora, cel puțin parțial, contextului secvențial local și/sau unei posibile presiuni de selecție naturală asupra exomului. În consecință, a fost aplicat un model de regresie liniară și am constatat că hipersensibilitatea DNAse 1, contextul insulelor CpG, valorile de conservare GERPP++ și nivelul de expresie au explicat ∼68-93% din rata DNM. Aceste constatări au indicat faptul că DNM-urile din exom s-au format independent de conservarea secvențelor ADN. Cu toate acestea, rata DNM a fost mai mare în genele ale căror produse erau nespecifice și în regiunile asemănătoare promotorilor activi din punct de vedere transcripțional.
În contrast cu rezultatele altor studii (Wong et al., 2016; Sandmann et al., 2017), am constatat că vârsta paternă nu a fost corelată cu rata DNM. Aceste constatări ar putea fi explicate prin faptul că setul de date a fost alcătuit din trio-uri cu vârste parentale similare și că a fost analizată doar o mică parte (∼1,5 %) din întregul genom. Pe baza acestor parametri, fiecare persoană a avut doar 1,9 (VarScan) sau 1,7 (VarSeqTM) DNM-uri în medie, comparativ cu 40-82 în întregul genom (Crow, 2000; Branciamore et al., 2010; Kong et al., 2012; Neale et al., 2012; Besenbacher et al., 2015; Francioli et al., 2015; Wong et al, 2016), în timp ce numărul de indels de novo în secvența de codificare a fost similar cu cel identificat în (Front Line Genomics, 2017).
Rezultatele analizei noastre funcționale extinse a adnotărilor au arătat că, din toate DNM-urile identificate, 4 (VarScan) și 35 (VarSeqTM) de variante au fost susceptibile de a fi DNM-uri patogene. Diferența în ceea ce privește numărul de DNM-uri patogene poate fi explicată prin faptul că, în funcție de algoritmul utilizat pentru identificarea DNM-urilor, ponderea DNM-urilor în secvențele codificatoare a fost semnificativ diferită. De exemplu, 21,05% dintre DNM-urile identificate de software-ul VarScan erau exonice, în timp ce 95,24% dintre cele identificate de software-ul VarSeqTM erau exonice. Aceste DNM-uri patogene se aflau în genele care codifică proteine esențiale pentru modelarea cromatinei, reglarea citoscheletului, modularea creșterii și vitalității celulare, funcția căilor de semnalizare citoplasmatică și inițierea răspunsului neuronal. În ciuda faptului că aceste DNM-uri sunt considerate patogene, toate persoanele care au participat la sondaj s-au identificat ca fiind „sănătoase” din punct de vedere genetic. Prin urmare, acest rezultat a indicat faptul că, în ciuda patogenității presupuse a DNM-urilor, genomurile în care erau localizate DNM-urile au tolerat în mod evident astfel de modificări, astfel încât manifestările bolii nu au fost adesea pronunțate. Potrivit lui Szamecz et al. (2014), cu cât DNM-urile apar mai des în poziții genetice conservate, cu atât mai puternice sunt efectele selecției naturale asupra modificărilor genetice prin mecanisme compensatorii de protecție a genomului. Efectele nocive ale variantelor pot fi atenuate în patru moduri. Unele gene pot tolera variantele trunchiate ale proteinelor, deoarece efectele funcționale ale acestora sunt mascate de expresia incompletă, de variantele compensatorii sau de semnificația funcțională scăzută a trunchierii (Bartha et al., 2015). În schimb, modificările genice asociate cu DNM-uri nesinonime sunt compensate prin mecanismul de acumulare a mutațiilor utile în întregul genom (Szamecz et al., 2014). Aceasta sugerează că, în aceste cazuri, mutațiile patogene nu sunt suficient de dăunătoare pentru a reduce fitness-ul mediu și, prin urmare, ele persistă mai mult timp în multe generații fiind modelate de selecția naturală.
În concluzie, analiza noastră a distribuției DNM-urilor și a contextului lor genetic și epigenetic a oferit informații despre variația genetică a genomului lituanian. Pe baza acestor constatări, studiile suplimentare în grupuri de pacienți cu boli genetice pot facilita capacitatea noastră de a distinge anumite DNM-uri patogene de DNM-urile de fond tolerate și de a identifica DNM-uri cauzale fiabile. Cu toate acestea, principala limitare a acestui studiu a constat în faptul că nu am examinat variația în regiunile genetice non-codificatoare și reglatoare. Aceste informații ar putea contribui la elucidarea posibilelor mecanisme de formare a DNM-urilor care rămân încă insuficient de clare.
Coduri de acces
Datele de secvență au fost depuse la Arhiva Europeană de Nucleotide (ENA), sub accesarea PRJEB25864 (ERP107829).
Declarație etică
Acest studiu a fost efectuat în conformitate cu recomandările de permisiune, Comitetul Regional de Etică pentru Cercetare Biomedicală din Vilnius. Protocolul a fost aprobat de Comitetul regional de etică pentru cercetare biomedicală din Vilnius. Toți subiecții și-au dat consimțământul informat în scris, în conformitate cu Declarația de la Helsinki.
Contribuții ale autorilor
LP a efectuat analiza datelor și a pregătit manuscrisul. AJ a calculat rata mutațiilor de novo. Secvențierea exomelor de trios a fost realizată de LA și IK. VK a fost cercetătorul principal.
Finanțare
Acest studiu a fost sprijinit de Fondul Social European în cadrul măsurii Global Grant. Proiectul LITGEN nr. VP1-3.1-ŠMM-07-K-01-013.
Declarație privind conflictul de interese
Autorii declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
Materiale suplimentare
Materialul suplimentar pentru acest articol poate fi găsit online la adresa: https://www.frontiersin.org/articles/10.3389/fgene.2018.00315/full#supplementary-material
Acuna-Hidalgo, R., Veltman, J. A., și Hoischen, A. (2016). Noi perspective în ceea ce privește generarea și rolul mutațiilor de novo în sănătate și boală. Genome Biol. 17:241. doi: 10.1186/s13059-016-1110-1
PubMed Abstract | Refef Full Text | Google Scholar
Bartha, I., Rausell, A., McLaren, P. J., Mohammadi, P., Tardaguila, M., Chaturvedi, N., et al. (2015). Caracteristicile variantelor heterozigote de trunchiere a proteinelor în genomul uman. PLoS Comput. Biol. 11:e1004647. doi: 10.1371/journal.pcbi.1004647
PubMed Abstract | CrossRef Full Text | Google Scholar
Besenbacher, S., Liu, S., Izarzugaza, J. M., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Variația nouă și ratele de mutație de novo în triourile daneze asamblate de novo la nivelul întregii populații. Nat Commun. 6:5969. doi: 10.1038/ncomms696969
PubMed Abstract | Refef Full Text | Google Scholar
Branciamore, S., Chen, Z. X., Riggs, A. D., și Rodin, S. R. (2010). Grupuri de insule CpG și selecție pro-epigenetică pentru CpG-uri în exoni codificatori de proteine ai HOX și ai altor factori de transcripție. Proc. Natl. acad. Sci. U.S.A. 107, 15485-15490. doi: 10.1073/pnas.1010506107
PubMed Abstract | CrossRef Full Text | Google Scholar
Butkiewicz, M., și Bush, W. S. (2016). Adnotarea funcțională in silico a variației genomice. Curr. Protoc. Hum. Genet. 88, 6.15.1-6.15.17.
Google Scholar
Crow, J. F. (2000). Originile, modelele și implicațiile mutațiilor spontane umane. Nat. Rev. Genet. 1, 40-47. doi: 10.1038/35049558
PubMed Abstract |Ref Full Text | Google Scholar
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., și Batzoglou, S. (2010). Identificarea unei fracțiuni mari din genomul uman care se află sub constrângere selectivă folosind GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
PubMed Abstract | Textul integral | Google Scholar
ENCODE Project Consortium (2012). O enciclopedie integrată a elementelor ADN din genomul uman. Nature 489, 57-74. doi: 10.1038/nature11247
PubMed Abstract | CrossRef Full Text | Google Scholar
Eyre-Walker, A., și Keightley, P. D. (2007). Distribuția efectelor de fitness ale noilor mutații. Nat. Rev. Genet. 8, 610-618. doi: 10.1038/nrg2146
PubMed Abstract |Ref Full Text | Google Scholar
Francioli, L. C., Polak, P. P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Modele și proprietăți la nivel de genom ale mutațiilor de novo la om. Nat. Genet. 47, 822-826. doi: 10.1038/ng.3292
PubMed Abstract | Refef Full Text | Google Scholar
Front Line Genomics (2017). Numărul 14 al revistei Front Line Genomics Magazine – ASHG. Londra: Front Line Genomics.
GTEx Consortium, Laboratory, Data Analysis andCoordinating Center (Ldacc)-Analysis Working Group., Statistical Methods groups-Analysis Working Group., Enhancing GTEx (eGTEx) groups, NIH Common et al. (2017). Efecte genetice asupra expresiei genice în țesuturile umane. Nature 550, 204-213. doi: 10.1038/nature24277
PubMed Abstract | Full CrossRef Text | Google Scholar
Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., et al. (2012). VarScan 2: descoperirea mutațiilor somatice și a modificărilor numărului de copii în cancer prin secvențierea exomului. Genome Res. 22, 568-576. doi: 10.1101/gr.129684.111
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rata mutațiilor de novo și importanța vârstei tatălui pentru riscul de boală. Nature 488, 471-475. doi: 10.1038/nature11396
PubMed Abstract | Full CrossRef Text | Google Scholar
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Másson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Diferențe la scară fină ale ratei de recombinare între sexe, populații și indivizi. Nature 467, 1099-1103. doi: 10.1038/nature09525
PubMed Abstract | Full Text | Google Scholar
Lappalainen, T., Sammeth, M., Friedlánder, M. R., ‘t Hoen, P. A., Monlong, J., Rivas, M. A., et al. (2013). Secvențierea transcriptomului și a genomului descoperă variații funcționale la om. Nature 501, 506-511. doi: 10.1038/nature12531
PubMed Abstract |Ref Full Text | Google Scholar
LITGEN (2011). Disponibil la: http://www.litgen.mf.vu.lt/
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242-245. doi: 10.1038/nature11011
PubMed Abstract | Ref. Full Text | Google Scholar
Peck, J. R., și Waxman, D. (2018). Ce este adaptarea și cum ar trebui să fie măsurată? J. Theor. Biol. 447, 190-198. doi: 10.1016/j.jtbi.2018.03.003
PubMed Abstract |Ref Full Text | Google Scholar
R Core Team (2013). Un limbaj și un mediu pentru calcul statistic. Viena: R Foundation for Statistical Computing.
Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
PubMed Abstract |Ref Full Text | Google Scholar
Sandmann, S., Graaf, A. O., de Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Nat. Sci. Rep. 7:43169. doi: 10.1038/srep43169
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S., și Sunyaev, S. (2008). Locurile nesinonime hipermutabile sunt supuse unei selecții negative mai puternice. PLoS Genet. 4:e1000281. doi: 10.1371/journal.pgen.1000281
PubMed Abstract | Full Text | Google Scholar
Subramanian, S., and Kumar, S. (2003). Substituțiile neutre apar cu o rată mai rapidă în exoni decât în ADN necodificator în genomurile de primate. Genome Res. 13, 838-844. doi: 10.1101/gr.1152803
PubMed Abstract | CrossRef Full Text | Google Scholar
Szamecz, B., Boross, G., Kalapis, D., Kovacs, K., Fekete, G., Farkas, Z., et al. (2014). The genomic landscape of compensatory evolution Be. The genomic landscape of compensatory evolution (Peisajul genomic al evoluției compensatorii). PLoS Biol. 12:e1001935. doi: 10.1371/journal.pbio.1001935
PubMed Abstract | CrossRef Full Text | Google Scholar
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). Baza de date STRING în 2017: rețele de asociere proteină-proteină de calitate controlată, făcute accesibile pe scară largă. Nucleic Acids Res. 45, D362-D368. doi: 10.1093/nar/gkw937
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, K., Li, M., și Hakonarson, H. (2010). ANNOVAR: adnotarea funcțională a variantelor genetice din datele de secvențiere de generație următoare. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
PubMed Abstract | CrossRef Full Text | Google Scholar
Warden, C. D., Adamson, A. W., Neuhausen, S. L., și Wu, X. (2014). Comparație detaliată a două pachete populare de apelare a variantelor pentru studiile exomului și ale exonului vizat. PeerJ 2:e600. doi: 10.7717/peerj.600
PubMed Abstract | Full Text | Google Scholar
Wong, W. S. W., Solomon, B. D. D., Bodian, D. L., Kothiyal, P., Eley, G., Huddleston, K. C., et al. (2016). Noi observații privind efectul vârstei materne asupra mutațiilor de novo din linia germinală. Nature communications 7:10486. doi: 10.1038/ncomms10486
PubMed Abstract | Text integral | Google Scholar
.