Incidența și prevalența calculilor renali continuă să crească în populația generală. Cu o prevalență pe parcursul vieții de până la 10%, nefrolitiaza (NL) și nefrocalcinoza (NC) reprezintă, prin urmare, sarcini de sănătate majore, în special în lumea occidentală (1). NL și NC sunt asociate cu o morbiditate semnificativă și cu progresia către boala cronică de rinichi din cauza recurenței, a intervențiilor chirurgicale/endoscopice repetitive și a inflamației concomitente. La un nivel simplificat, formarea calculilor renali rezultă dintr-un dezechilibru al inhibitorilor urinari (de exemplu, citrat, magneziu, uromodulină și pirofosfat) și al promotorilor (de exemplu, oxalat, calciu, fosfat, urat și cistină) de cristalizare, depășind suprasaturația cu agregare consecutivă, nucleare și creștere a calculilor la nivelul plăcii lui Randall (figura 1). Acest dezechilibru se poate datora unei manipulări enterale și/sau renale alterate fie de promotori, fie de inhibitori, cum ar fi malsecreția enterală de oxalat sau malreabsorbția renală de calciu (figura 1).

Figura 1. Dezechilibrul inhibitorilor și promotorilor urinari ai cristalizării care duc la formarea calculilor renali. Concentrația inhibitorilor și a promotorilor urinari este influențată și controlată atât de transportatorii intestinali, cât și de cei renali (axa GI-rein). Acești transportatori și schimbători, cum ar fi SLC26A1, sunt responsabili pentru secreție și absorbție. În ceea ce privește oxalatul, hiperabsorbția enterală, dar malsecreția, și/sau hipersecreția renală, dar malabsorbția duce la niveluri de oxalat urinar care depășesc suprasaturația și, astfel, favorizează cristalizarea și formarea consecutivă de calculi .
Etiologia care stă la baza NL este considerată a fi multifactorială, cu o componentă de mediu, dietetică notabilă, hormonală și genetică. În studiile gemelare, heritabilitatea calculilor renali a fost estimată la 56% (3), iar până la două treimi dintre cei care formează calculi hipercalciurici au rude cu NL (4). Deși calculii renali cu conținut de calciu reprezintă peste 80% din totalul acestora, baza genetică a acestor calculi rămâne în mare parte necunoscută (5). Cu excepția variantelor din CLDN14, TRPV5, SLC34A1, ALPL, CASR și UMOD, studiile de asociere la nivel de genom încă nu au dat naștere unor factori genetici substanțiale (6-8). Cu toate acestea, au fost identificate alele de risc în cadrul unor gene despre care s-a constatat, de asemenea, că transmit boala pe o bază mendeliană, cum ar fi CASR, SLC34A1 și SLC2A9 (9, 10). Până în prezent, au fost identificate mai mult de 30 de gene unice cu un fenotip definit de moștenirea mendeliană online la om care sunt implicate în NL/NC, dacă sunt mutate (Tabelul 1).

Tabel 1. Gene, cunoscute ca fiind cauza formelor monogenice de NL/NC.
Modele de moștenire în formele monogenice includ transmiterea autosomal-dominantă, autosomal-recesivă și X-linked. În mod interesant, la câteva dintre aceste gene, au fost raportate atât moduri de moștenire recesivă, cât și dominantă: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12 și SLC4A1. În timp ce cele mai multe dintre tulburările congenitale sindromice și severe prezintă un model de moștenire recesivă (Bartter, Lowe, Dent, FHHNC și acidoza tubulară renală distală cu surditate neurosenzorială), afecțiunile mai ușoare sunt mai degrabă asociate cu mutații în genele dominante. Majoritatea proteinelor codificate constituie transportatori renali de solut (de exemplu, SLC34A1, SLC34A3 și SLC9A3R1), dar și canale de clorură (CLCN5), proteine de joncțiune strânsă (de exemplu, CLDN16/CLDN19) și enzime metabolizatoare (de exemplu, AGXT, APRT și CYP24A1) au fost găsite defecte la pacienții cu NL/NC. Prin urmare, defectul de bază este localizat în cea mai mare parte în sistemul tubular al rinichiului propriu-zis și, prin urmare, poate fi atribuit ca tubulopatie. În schimb, afecțiuni a priori extrarenale, ca în cazul hiperoxaluriei primare (PH), în care disfuncția enzimelor hepatice (AGXT, GRHPR și HOGA1) determină acumularea de oxalat cu afectare renală secundară, sunt cauze conceptibile de NL/NC. Deși se consideră că fiecare fenotip de boală reprezintă o entitate relativ rară, cauzele monogenice pot reprezenta un număr semnificativ de pacienți prin eterogenitatea lor genetică largă (42). În afară de eterogenitatea genetică, există, de asemenea, o variație alelică, în care variantele trunchiate determină mai degrabă o pierdere de funcție, iar variantele missense (hipomorfe) pot cauza defecte mai degrabă subtile, care pot fi supravegheate clinic, în special la adulții care formează calculi. Un alt fenomen apreciat recent se referă la efectele dozării genice în mai multe dintre genele de calculi renali menționate anterior. În cazul SLC34A3, de exemplu, care codifică unul dintre principalii transportatori de fosfat din tubulul proximal (NaPiIIc), s-a demonstrat că indivizii heterozigoți nu mai pot fi considerați pur și simplu purtători sănătoși, deoarece aceștia prezintă calcificări renale și/sau manifestări osoase semnificativ mai frecvente decât indivizii de tip sălbatic; dar totuși într-o măsură mai mică decât indivizii bialelici (homozigoți și heterozigoți compuși) (43). Observații similare au fost raportate pentru familiile cu mutații în CYP24A1 (44). Contribuția tulburărilor monogenice la prevalența generală a calculilor renali nu a fost studiată în mod exhaustiv în trecut. În special, lipsesc dovezile genetice bazate pe depistări ample ale unei multitudini de gene cauzatoare în cohorte mari de pacienți. Testarea genetică cuprinzătoare a fost prea costisitoare și ineficientă în trecut. Pentru majoritatea persoanelor cu NL/NC, analiza mutațiilor pentru un defect genetic cauzal nu a fost, prin urmare, accesibilă, în ciuda faptului că cunoașterea cauzei moleculare a NL/NC poate avea consecințe importante pentru prognostic, profilaxie și/sau tratament. Doar estimări aproximative au fost derivate din studiile de observație clinică: pe baza unei colecții uriașe de date de analiză a compoziției pietrelor, s-a ajuns la concluzia că cauzele monogenice nu depășesc 9,6 % la copii și 1,6 % la adulți (45). Cu toate acestea, în ultimul deceniu, această situație a început să se schimbe, odată cu apariția tehnicilor de secvențiere de mare randament.
Analiză de mutații de mare randament la pacienții cu NL/NC
Pentru a investiga pacienții cu calculi renali în vederea depistării prezenței mutațiilor patogene în genele cunoscute ale bolii, am stabilit un panel de gene bazat pe multiplex-PCR microfluidică și secvențiere NextGenen consecutivă (Fluidigm™/NGS) (46, 47).
În cadrul unui „studiu-pilot”, am recrutat consecutiv 268 de persoane nerezolvate genetic din clinicile tipice de calculi renali; 102 probanzi pediatrici și 166 adulți. Ca rezultat, am identificat 50 de variante dăunătoare în 14 din cele 30 de gene analizate, ceea ce a condus la un diagnostic molecular în 15% din toate cazurile. În subgrupul pediatric, am detectat o mutație cauzală în 21 %, în timp ce în rândul adulților, variantele deletere au fost prezente în 11 % (Figura 2A) (48). Mutațiile în gena SLC7A9 a cistinuriei au fost găsite cel mai frecvent în cohorta de adulți (Figura 2B). Două studii de urmărire au reușit să confirme aceste rezultate. În primul rând, într-o cohortă exclusiv pediatrică de 143 de pacienți cu NL/NC, 17% din cazuri au fost explicate prin mutații în 14 gene diferite (49). În al doilea rând, într-o cohortă de 51 de familii cu vârsta de manifestare a NL/NC înainte de 25 de ani, WES țintit a fost utilizat pentru a detecta o cauză genetică în aproape 30% (50). Nu este surprinzător faptul că mutațiile recesive au fost găsite mai frecvent în rândul nou-născuților și în cazurile de boală congenitală, în timp ce afecțiunile dominante s-au manifestat de obicei mai târziu în viață. Aceste date indică faptul că boala genetică a calculilor renali este o afecțiune subdiagnosticată, în ciuda faptului că diagnosticul molecular va influența potențial prognosticul, profilaxia și/sau tratamentul. Cu toate acestea, o limitare care merită menționată este o potențială prejudecată de selecție datorată recrutării din clinicile specializate în calculi renali în toate cele trei studii menționate mai sus.

Figura 2. Analiza mutațiilor a 30 de gene cunoscute ale nefrolitiazei monogenice (NL)/nefrocalcinozei (NC) la 268 de pacienți cu NL/NC. (A) Fracția de cauze monogenice în subcohorta pediatrică și adultă. (B) Numărul de cauze monogenice pe gene (roșu denotă adulții; portocaliu denotă pacienții pediatrici). De remarcat că SLC7A9 a fost găsită cel mai frecvent mutantă, în special la adulții tineri.
Identificarea noilor gene ale bolilor umane prin abordarea genelor candidate
Analiza mutațiilor de mare randament este, de asemenea, utilizată pentru a depista variantele patogene în diferite gene candidate. Una dintre cele mai interesante descoperiri recente a fost descoperirea de mutații umane în SLC26A1 (32). De la prima descriere a formării de calculi renali de Ca-oxalat (CaOx) și a NC la șoarecii SLC26a1 (Sat1)-knockout de către Dawson et al. în 2010, SLC26A1 a fost o genă candidată de bună credință la NL (51). SLC26A1 codifică un schimbător de anioni exprimat la nivelul membranei bazolaterale a tubulilor renali proximali, a ileonului și a jejunului. În consecință, prin utilizarea unei abordări de genă-candidat, au fost identificate variante patogene la oamenii cu antecedente de CaOx-NL cu debut precoce, și anume la doi indivizi neînrudiți cu variante bialelice missense (32). Din punct de vedere funcțional, patogenitatea variantelor identificate a fost demonstrată in vitro, conducând la un trafic intracelular greșit și la o activitate de transport afectată (32). Defectul SLC26A1 constituie, prin urmare, o nouă cauză a CaOx-NL și ar trebui luată în considerare atunci când se testează indivizii pentru cauzele formării recurente de calculi de CaOx.
Noua evidență clinică a pacienților pentru boala ereditară a calculilor renali
Majoritatea datelor epidemiologice privind creșterea prevalenței în țările occidentale provin din bazele de date din SUA. Deși sunt necesare de urgență, bazele de date europene centralizate nu sunt disponibile în acest moment. Deoarece studiile genetice menționate anterior privind prevalența patologiei ereditare a calculilor renali au fost executate cu cohorte mici din centre specializate atât din Europa, cât și din SUA, o transpunere la situația generală din Europa nu este valabilă. În timp ce în SUA, Rare Kidney Stone Consortium constituie o platformă care integrează și coordonează activitățile de registru, de știință fundamentală și de cercetare clinică pentru afecțiuni rare, cum ar fi cistinuria, PH, deficitul de APRT, boala Dent și Lowe, în prezent, nici în Europa și nici în Germania nu a fost pusă în aplicare o colectare de date comparabilă cu privire la pacienții cu boli ereditare ale calculilor renali. În colaborare cu registrul european existent al PH, OxalEurope (Prof. Bernd Hoppe, Universitatea din Bonn), și prin finanțare din partea Deutsche Forschungsgemeinschaft și Else Kröner-Fresenius Stiftung, am înființat recent un „Registru al pacienților clinici pentru boala ereditară a calculilor renali” la Universitatea din Leipzig. Registrul este susținut la nivel național de Societățile germane de nefrologie pentru adulți (DGfN) și de nefrologie pediatrică (GPN). Acesta este înscris în continuare în Registrul german de studii clinice (DRKS-ID: DRKS00012891). Ca parte fundamentală a recrutării studiului, analiza mutațiilor de mare capacitate pentru genele cunoscute și noi ale calculilor renali este oferită pe bază de cercetare pentru pacienții fără un diagnostic molecular stabilit, dar cu un tablou clinic care indică o susceptibilitate genetică subiacentă: de exemplu, vârsta precoce de debut (<40 de ani), antecedente familiale pozitive, fenotipuri indicative, cum ar fi NC, cistinurie sau RTA, și NL sever recurentă (>3×) (tabelul 2) (tabelul 2). În timp ce pacienții cu un diagnostic genetic deja stabilit sunt în general înrolați, cazurile cu cauze secundare de NL/NC, cum ar fi malignitatea, sarcoidoza și hiperparatiroidismul primar, nu sunt incluse în analiza genetică. Pentru a înrola în mod activ pacienți, un centru clinic va avea nevoie, de obicei, de aprobarea din partea Comitetului de evaluare instituțională local; un proces pentru care noi ne oferim ajutorul și asistența prin furnizarea modelelor respective. În urma aprobării etice, formularul de consimțământ și fișele de date clinice (de exemplu, chestionarul clinic) pot fi descărcate de pe site-ul web al registrului nostru (http://www.mks-registry.net). Pentru a asigura o fenotipare clinică amănunțită, vom solicita informații substanțiale despre pacienți, cum ar fi etnia, consangvinitatea, istoricul familial, vârsta de debut, recurența (definită ca fiecare eveniment presupus a fi un nou calcul renal), aportul zilnic de lichide, intervențiile chirurgicale și implicarea extrarenală, printre altele. Documentele pot fi completate de către pacient cu ajutorul medicului care îl înscrie. În plus, parametrii serici biochimici, inclusiv creatinina, eGFR, PTH, vitamina D, electroliții, acidul uric și analiza urinei (pH, calciu, fosfat, magneziu, acid uric, citrat, oxalat și cistină, de preferință din urina de 24 de ore, dacă nu din urină punctiformă), precum și date privind analiza compoziției calculilor vor fi solicitate la înscriere. Ținând cont de faptul că recoltarea urinei de 24 de ore și analiza compoziției calculilor nu se efectuează în mod obișnuit în toate instituțiile, includem acești parametri în funcție de disponibilitate. Vizitele de monitorizare clinică la 2 ani a pacienților înscriși sunt de dorit, dar nu obligatorii. După înregistrare, centrele clinice care recrutează vor primi un login personalizat pentru a introduce datele pacienților prin intermediul site-ului nostru de înregistrare (http://www.mks-registry.net). Alternativ, oferim introducerea datelor în format electronic atunci când ne sunt trimise pe hârtie. Datele introduse vor fi stocate pe un server securizat și pot fi accesate de către centrele clinice participante pentru a vizualiza propriile date despre pacienți. Următoarele site-uri web oferă informații suplimentare:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891
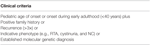
Tabel 2. Criterii de includere pentru analiza mutațiilor în registrul clinic de pacienți.
În rezumat, boala calculilor renali este o afecțiune din ce în ce mai răspândită, care este heterogenă din punct de vedere clinic și slab înțeleasă, în special factorii genetici care o determină. După cum a indicat o serie de studii recente, afecțiunile monogenice sunt cel mai probabil subestimate în ceea ce privește prevalența. Prin punerea în aplicare a unui registru centralizat al pacienților cu privire la boala ereditară a calculilor renali, vom contribui la depășirea, cel puțin în parte, a marelui deficit de cunoștințe privind genetica calculilor renali. În acest context, registrele clinice sunt surse valoroase din mai multe motive: în primul rând, delimitarea unor mai bune asocieri fenotip-genotip va fi crucială pentru o stratificare mai precisă a pacienților în viitoarele studii de cercetare clinică. În al doilea rând, identificarea de noi gene de boală cu noi mecanisme de boală va diminua decalajul etiologiei necunoscute a NL/NC; și în al treilea rând, descifrarea de noi ținte moleculare ajută la deschiderea drumului pentru dezvoltarea de medicamente de prevenire a recidivelor în familiile grav afectate.
Declarație de etică
Acest studiu a fost efectuat în conformitate cu recomandările „Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” cu consimțământul informat scris al tuturor subiecților. Toți subiecții și-au dat consimțământul informat în scris în conformitate cu Declarația de la Helsinki. Protocolul a fost aprobat de „Ethikkommission an der Medizinischen Fakultät der Universität Leipzig.”
Contribuții ale autorilor
JH a conceput și a scris manuscrisul. BH, AS, LM și RS au editat manuscrisul și au construit infrastructura registrului care este prezentată cititorului.
Declarație privind conflictul de interese
Autorii declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
Finanțare
Cercetarea este finanțată prin granturi de proiect de la DFG (HA 6908/2-1) și EKFS (2016_A52) pentru JH. Această lucrare a fost susținută în continuare de Ministerul Federal al Educației și Cercetării (BMBF), Germania, FKZ: 01EO1501 către JH.
1. Scales CD Jr., Smith AC, Hanley JM, Saigal CS. Prevalența pietrelor la rinichi în Statele Unite. Eur Urol (2012) 62:160-5.
Google Scholar
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973-85. doi:10.1053/j.ajkd.2016.05.016
CrossRef Full Text | Google Scholar
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053-61. doi:10.1111/j.1523-1755.2005.00170.x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Stechman MJ, Loh NY, Thakker RV. Genetica nefrolitiazei hipercalciurice: boala pietrelor renale. Ann N Y Acad Sci (2007) 1116:461-84. doi:10.1196/annals.1402.030
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Sayer JA. Progrese în înțelegerea geneticii nefrolitiazei cu conținut de calciu. J Am Soc Nephrol (2017) 28:748-59. doi:10.1681/ASN.2016050576.
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Variantele de secvență în gena CLDN14 se asociază cu calculii renali și densitatea minerală osoasă. Nat Genet (2009) 41:926-30. doi:10.1038/ng.404
PubMed Abstract | Full Text | Google Scholar
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
CrossRef Full Text | Google Scholar
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 influențează concentrațiile de acid uric cu efecte pronunțate specifice sexului. Nat Genet (2008) 40:430-6. doi:10.1038/ng.107
PubMed Abstract | Text integral | Google Scholar
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutațiile în gena transportatorului de glucoză 9 SLC2A9 cauzează hipouricemie renală. Am J Hum Genet (2008) 83:744-51. doi:10.1016/j.ajhg.2008.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identificarea și caracterizarea unei gene cu substituții de baze asociate cu fenotipul de hipercalciurie absorbantă și densitatea osoasă spinală scăzută. J Clin Endocrinol Metab (2002) 87:1476-85. doi:10.1210/jcem.87.4.8300
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Direcționarea necorespunzătoare a L-alaninei L-alanină:glioxilat aminotransferazei peroxisomale către mitocondrie la pacienții cu hiperoxalurie primară depinde de activarea unei secvențe criptice de direcționare mitocondrială printr-o mutație punctiformă. Proc Natl Acad Sci U S A (1991) 88:10900-4. doi:10.1073/pnas.88.23.10900
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Adenină fosforibosiltransferaza umană. Identificarea mutațiilor alelice la nivel de nucleotide ca o cauză a deficienței complete a enzimei. J Clin Invest (1987) 80:1409-15. doi:10.1172/JCI113219
CrossRef Full Text | Google Scholar
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutațiile în ATP6N1B, care codifică o nouă subunitate 116-kD a pompei de protoni vacuolare renale, cauzează acidoză tubulară renală distală recesivă cu auz conservat. Nat Genet (2000) 26:71-5. doi:10.1038/79208
PubMed Abstract | Text integral | Google Scholar
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutațiile în gena care codifică subunitatea B1 a H+-ATPazei provoacă acidoză tubulară renală cu surditate neurosenzorială. Nat Genet (1999) 21:84-90. doi:10.1038/5022
PubMed Abstract | Text integral | Google Scholar
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Sindromul de deficit de anhidrază carbonică II într-o familie belgiană este cauzat de o mutație punctiformă la un reziduu invariant de histidină (107 His→Tyr): structura completă a genei CA II umane normale. Am J Hum Genet (1991) 49:1082-90.
Google Scholar
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. Un sindrom familial de hipocalcemie cu hipercalciurie datorat unor mutații în receptorul de detectare a calciului. N Engl J Med (1996) 335:1115-22. doi:10.1056/NEJM199610103351505
PubMed Abstract | Reflect Full Text | Google Scholar
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. O bază moleculară comună pentru trei boli moștenite ale calculilor renali. Nature (1996) 379:445-9. doi:10.1038/379445a0
PubMed Abstract | Full Text | Google Scholar
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutațiile în gena canalului de clorură, CLCNKB, cauzează sindromul Bartter de tip III. Nat Genet (1997) 17:171-8. doi:10.1038/ng1097-171
CrossRef Full Text | Google Scholar
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, o proteină de joncțiune strânsă renală necesară pentru resorbția paracelulară de Mg2+. Science (1999) 285:103-6. doi:10.1126/science.285.5424.103
PubMed Abstract | Refef Full Text | Google Scholar
21. Konrad M, Schaller A, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutațiile în gena de joncțiune strânsă claudin 19 (CLDN19) sunt asociate cu deficitul renal de magneziu, insuficiență renală și afectare oculară severă. Am J Hum Genet (2006) 79:949-57. doi:10.1086/508617
PubMed Abstract | Refef Full Text | Google Scholar
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutații în CYP24A1 și hipercalcemia infantilă idiopatică. N Engl J Med (2011) 365:410-21. doi:10.1056/NEJMoa1103864
CrossRef Full Text | Google Scholar
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nefrocalcinoză (sindromul renal de smalț) cauzată de mutații autosomal recesive FAM20A. Nephron Physiol (2012) 122:1-6. doi:10.1159/000349989
PubMed Abstract | Red Cross Full Text | Google Scholar
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. Gena care codifică hidroxipiruvat reductaza (GRHPR) este mutantă la pacienții cu hiperoxalurie primară de tip II. Hum Mol Genet (1999) 8:2063-9. doi:10.1093/hmg/8.11.2063
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hamilton AJ, Bingham C, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. Mutația HNF4A R76W cauzează sindromul Fanconi dominant atipic în plus față de un fenotip al celulelor β. J Med Genet (2014) 51:165-9. doi:10.1136/jmedgenet-2013-102066
CrossRef Full Text | Google Scholar
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutațiile în DHDPSL sunt responsabile de hiperoxaluria primară de tip III. Am J Hum Genet (2010) 87:392-9. doi:10.1016/j.ajhg.2010.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identificarea a 17 mutații independente responsabile de deficitul de hipoxantină-guanină fosforibosiltransferază umană (HPRT). Am J Hum Genet (1991) 48:951-8.
PubMed Abstract | Google Scholar
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Eterogenitatea genetică a sindromului Bartter dezvăluită prin mutații în canalul K+, ROMK. Nat Genet (1996) 14:152-6. doi:10.1038/ng1096-152
CrossRef Full Text | Google Scholar
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, sindrom Bartter’s transient antenatal și mutații MAGED2. N Engl J Med (2016) 374:1853-63. doi:10.1056/NEJMoa1507629
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Markerii flanking strâns legați pentru sindromul Lowe oculocerebrorenal, cu aplicare la evaluarea purtătorilor. Am J Hum Genet (1988) 42:748-55.
PubMed Abstract | Google Scholar
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Sindromul Bartter, alcaloză hipocaliemică cu hipercalciurie, este cauzat de mutații în cotransportatorul Na-K-2Cl NKCC2. Nat Genet (1996) 13:183-8. doi:10.1038/ng0696-183
PubMed Abstract | Textul integral | Google Scholar
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutațiile în SLC26A1 cauzează nefrolitiază. Am J Hum Genet (2016) 98:1228-34. doi:10.1016/j.ajhg.2016.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Identificarea moleculară a unui schimbător renal de anioni de urat care reglează nivelul de urat din sânge. Nature (2002) 417:447-52. doi:10.1038/nature742
PubMed Abstract | Full CrossRef Text | Google Scholar
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nefrolitiază și osteoporoză asociate cu hipofosfatemia cauzată de mutații în cotransportatorul sodiu-fosfat de tip 2a. N Engl J Med (2002) 347:983-91. doi:10.1056/NEJMoa020028
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet (2006) 78:193-201. doi:10.1086/499410
PubMed Abstract | Text integral | Google Scholar
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cistinuria cauzată de mutații în rBAT, o genă implicată în transportul de cistină. Nat Genet (1994) 6:420-5. doi:10.1038/ng0494-420
PubMed Abstract | Refef Full Text | Google Scholar
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest (1997) 100:1693-707. doi:10.1172/JCI119694
CrossRef Full Text | Google Scholar
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet (1999) 23:52-7. doi:10.1038/12652
PubMed Abstract | Text integral | Google Scholar
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med (2008) 359:1128-35. doi:10.1056/NEJMoa0802836
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Scott P, Ouimet D, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Dovezi sugestive pentru o genă de susceptibilitate în apropierea locusului receptorului de vitamina D în formarea idiopatică a calculilor de calciu. J Am Soc Nephrol (1999) 10:1007-13.
PubMed Abstract | Google Scholar
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identificarea a două mutații în gena xantină dehidrogenazei umane responsabile de xantinuria clasică de tip I. J Clin Invest (1997) 99:2391-7. doi:10.1172/JCI119421
PubMed Abstract | Full CrossRef Text | Google Scholar
42. Hildebrandt F. Bolile genetice ale rinichiului. Lancet (2010) 375:1287-95. doi:10.1016/S0140-6736(10)60236-X
CrossRef Full Text | Google Scholar
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutațiile în SLC34A3/NPT2c sunt asociate cu calculi renali și nefrocalcinoză. J Am Soc Nephrol (2014) 25:2366-75. doi:10.1681/ASN.2013101085
PubMed Abstract | CrossRef Full Text | Google Scholar
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Fenotipuri clinice și biochimice ale adulților cu mutații monoalelice și bialelice ale CYP24A1: dovezi ale efectului dozei genei. Osteoporos Int (2016) 27:3121-5. doi:10.1007/s00198-016-3615-6
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Récents acquis diagnostiques et thérapeutiques. Nephrol Ther (2008) 4:231-55. doi:10.1016/j.nephro.2007.12.005
CrossRef Full Text | Google Scholar
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49:756-67. doi:10.1136/jmedgenet-2012-100973
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identificarea a 99 de noi mutații într-o cohortă mondială de 1.056 de pacienți cu o ciliopatie legată de nefronoftizie. Hum Genet (2013) 132:865-84. doi:10.1007/s00439-013-1297-0
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Paisprezece gene monogenice reprezintă 15% din nefrolitiază/nefrocalcinoză. J Am Soc Nephrol (2015) 26:543-51. doi:10.1681/ASN.2014040388
CrossRef Full Text | Google Scholar
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalența cauzelor monogenice la pacienții pediatrici cu nefrolitiază sau nefrocalcinoză. Clin J Am Soc Nephrol (2016) 11:664-72. doi:10.2215/CJN.07540715
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Secvențierea întregului exom detectează frecvent o cauză monogenică în nefrolitiaza și nefrocalcinoza cu debut precoce. Kidney Int (2018) 93:204-13. doi:10.1016/j.kint.2017.06.025
CrossRef Full Text | Google Scholar
51. Dawson PA, Russell CS, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolitiaza și hepatotoxicitatea sunt legate de transportatorul de anioni Sat1 la șoareci. J Clin Invest (2010) 120:706-12. doi:10.1172/JCI31474
PubMed Abstract | CrossRef Full Text | Google Scholar
.