Alexei Drummond, Andrew Rambaut, Remco Bouckaert, and Walter Xie
1 Introducere
Acest tutorial prezintă software-ul BEAST pentru analiza evolutivă bayesiană printr-un tutorial simplu. Tutorialul implică co-estimarea unei filogenii genetice și a timpilor de divergență asociați în prezența informațiilor de calibrare din dovezile fosile.
Aveți nevoie de următorul software la dispoziție:
- BEAST – acest pachet conține programul BEAST, BEAUti, TreeAnnotator și alte programe utilitare. Acest tutorial este scris pentru BEAST v2.2.x, care are suport pentru partiții multiple. Este disponibil pentru descărcare de la http://www.beast2.org/.
- Tracer – acest program este utilizat pentru a explora rezultatul programului BEAST (și al altor programe MCMC bayesiene). Acesta rezumă grafic și cantitativ distribuțiile parametrilor continui și oferă informații de diagnosticare. La momentul redactării, versiunea actuală este v1.6. Este disponibil pentru descărcare de la
http://tree.bio.ed.ac.uk/software/. - FigTree – aceasta este o aplicație pentru afișarea și imprimarea filologiilor moleculare, în special a celor obținute cu ajutorulBEAST. La momentul scrierii acestui articol, versiunea actuală este v1.4.2. Este disponibilă pentru descărcare de la http://tree.bio.ed.ac.uk/software/.
Acest tutorial vă va ghida în analiza unei alinieri de secvențe prelevate din douăsprezece specii de primate (a se vedea figura 1). Scopul este de a estima filogenia, rata de evoluție pe fiecare descendență și vârstele divergențelor necalibrateancestrale.
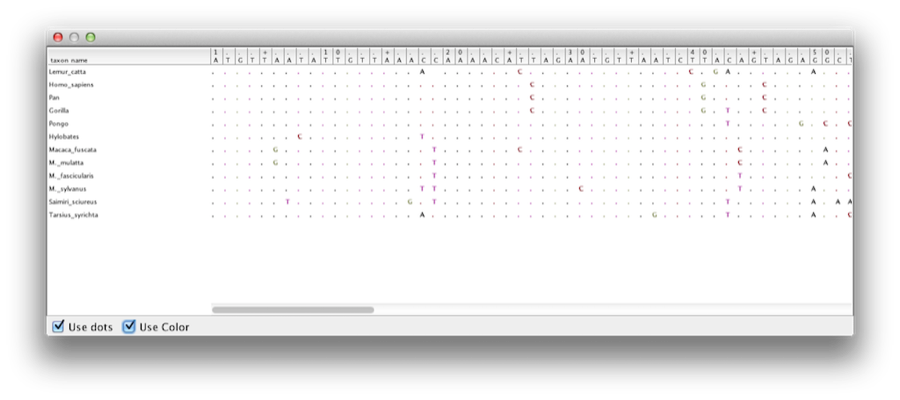
Figura 1: O parte din alinierea pentru primate.
Primul pas va fi convertirea unui fișier NEXUS cu un bloc DATA sau CHARACTERS într-un fișier de intrare BEAST XML. Acest lucru se face cu ajutorul programului BEAUti (care înseamnă Bayesian Evolutionary Analysis Utility). Acesta este un program ușor de utilizat pentru setarea modelului evolutiv și a opțiunilor pentru analiza MCMC. Al doilea pas este de a rula efectiv BEAST folosind fișierul de intrare generat de BEAUTi, careconține datele, modelul și setările de analiză. Pasul final constă în explorarea ieșirii BEAST pentru a diagnostica problemele și pentru a rezuma rezultatele.
2 BEAUti
Programul BEAUti este un program ușor de utilizat pentru stabilirea parametrilor modelului pentru BEAST. Rulați BEAUti făcând dublu clic pe pictograma sa. Odată rulat, BEAUti va avea un aspect similar, indiferent de sistemul de calcul pe care este rulat. Pentru acest tutorial, în figuri este utilizată versiunea Mac OS X, dar versiunile Linux și Windows vor avea aceeași dispunere și funcționalitate.
2.1 Încărcarea fișierului NEXUS
Pentru a încărca o aliniere în format NEXUS, selectați pur și simplu opțiunea Import Alignment… din meniul File sau trageți fișierul în mijlocul panoului Partitions.
Fila de exemplu numită primate-mtDNA.nex este disponibilă din directorul examples/nexus/ pentru Mac și Linux și examples/nexus/ pentru Windows în interiorul directorului în care a fost instalat BEAST. acest fișier conține o aliniere a secvențelor a 12 specii de primate.
O fereastră de adăugare a unei partiții (Figura 2) se va deschide dacă pachetul aferent este instalat. Dacă utilizați BEAST 2 „pur”, puteți trece la paragraful următor. În caz contrar, selectați Add Alignment (Adăugare aliniere) și faceți clic pe OK pentru a continua.
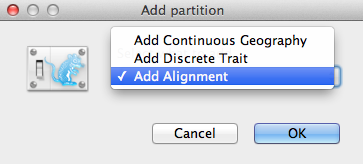
Figura 2: Fereastra Add Partition (Adăugare partiție) (Apare numai dacă sunt instalate pachetele conexe).
Dacă există suprapuneri de codificare în partiții, va apărea fereastra cu mesaj de avertizare (Figura 3). Citiți și faceți clic pe OK pentru a continua.
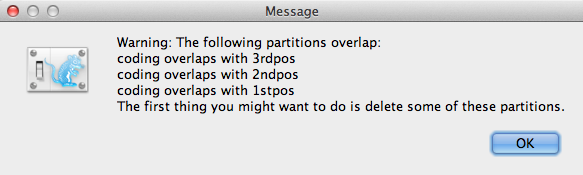
Figura 3: Fereastra cu mesaj de avertizare (apare numai dacă există suprapuneri de codificare în partiții).
După ce au fost încărcate, în panoul principal sunt afișate cinci partiții de caractere (Figura 4). Alinierea este împărțită într-o parte codificatoare de proteine și o parte necodificatoare,iar partea codificatoare este împărțită în pozițiile de codon 1, 2 și 3. Trebuie să eliminați partiția „coding” înainte de a trece la pasul următor, deoarece aceasta se referă la aceleași nucleotide ca și partițiile „1stpos”, „2ndpos” și „3rdpos”. Pentru a elimina partiția „coding”, selectați rândul și faceți clic pe butonul „-” din partea de jos a tabelului. Puteți vizualiza alinierea dând dublu clic pe partiție.
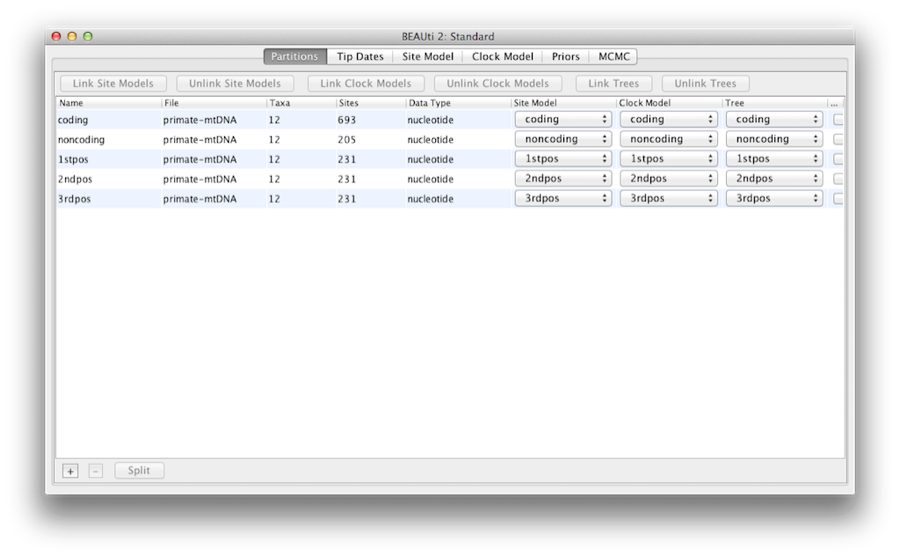
Figura 4: O captură de ecran a filei de date din BEAUti. Aceasta și toate capturile de ecran următoare au fost realizate pe un computer Apple care rulează Mac OS X și vor avea un aspect ușor diferit pe alte sisteme de operare.
Link/Unlink partition models
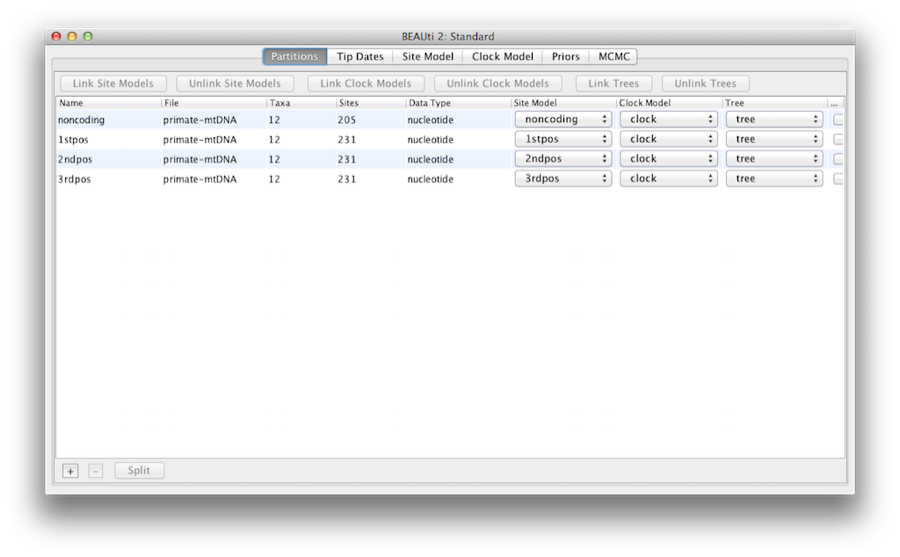
Figura 5: O captură de ecran a filei Partitions (Partiții) din BEAUti după ce a fost legat și redenumit modelul de ceas și arborele.
Din moment ce secvențele sunt legate (adică provin toate din genomul mitocondrial, despre care se crede că nu suferă recombinare la păsări și mamifere), ele au aceeași ascendență, astfel încât partițiile ar trebui să aibă același arbore temporal în model. Din motive de simplitate, vom presupune, de asemenea, că partițiile au aceeași rată de evoluție pentru fiecare ramură și, prin urmare, același „model de ceas”.Ne vom limita modelarea eterogenității ratei la eterogenitatea între situsuri în cadrul fiecărei partiții și vom permite, de asemenea, ca partițiile să aibă rate medii de evoluție diferite.
Deci, în acest moment va trebui să legăm modelul de ceas și arborele. În panoul Partitions, selectați toate cele patru partiții din tabel (sau niciuna, în mod implicit toate partițiile sunt afectate) și faceți clic pe butonul Link Trees (Legătura arborilor) și apoi pe butonul Link Clock Models (Legătura modelelor de ceas) (a se vedea figura 5). Apoi faceți clic pe primul meniu derulant din coloana Clock Model și redenumiți modelul de ceas partajat în „clock”. În mod similar, redenumiți arborele partajat în „tree”. Acest lucru va face ca următoarele opțiuni și fișierele jurnal generate să fie mai ușor de citit.
2.2 Setarea modelului de substituție
Postul următor este de a configura modelul de substituție. Apoi, selectați fila Site Models din partea de sus a ferestrei principale (sărim peste fila Tip Dates, deoarece toți taxonii provin din eșantioane contemporane). Acest lucru va dezvălui setările modelului evolutiv pentru BEAST. Opțiunile disponibile depind de faptul dacă datele sunt nucleotide, sau aminoacizi, date binare sau date generale. Setările care vor apărea după încărcarea alinierii nucleotidelor de primate vor fi valorile implicite pentru datele de nucleotide, așa că trebuie să facem câteva modificări.
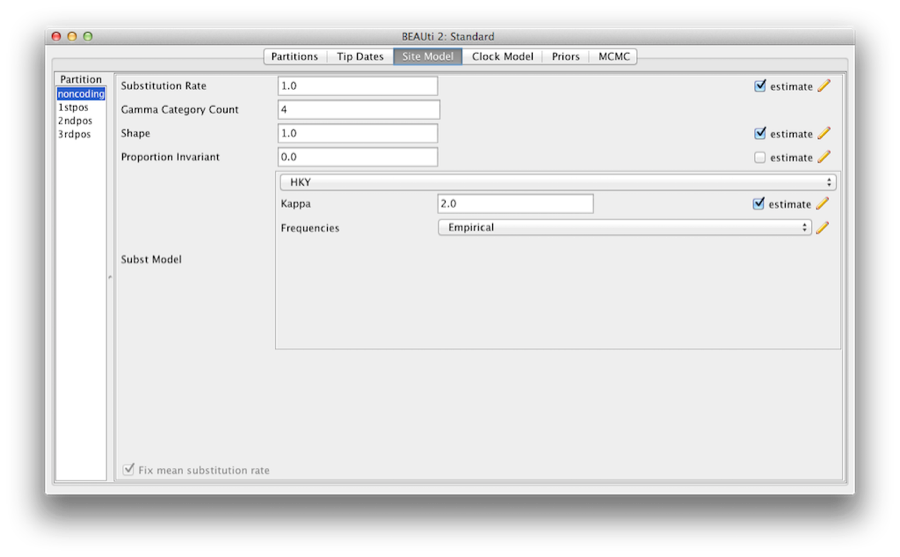
Figura 6: O captură de ecran a filei modelului de site din BEAUti.
Majoritatea modelelor ar trebui să vă fie familiare. Mai întâi, setați numărul de categorii Gamma la 4 și apoi bifați căsuța „estimate” pentru parametrul Shape. Acest lucru va permite modelarea variației ratei între siturile din fiecare partiție. Rețineți că 4 până la 6 categorii funcționează suficient de bine pentru majoritatea seturilor de date, în timp ce un număr mai mare de categorii necesită mai mult timp de calcul pentru un beneficiu suplimentar redus. Lăsăm intrarea Proportion Invariant (Proporție invariantă) setată la zero.
Apoi selectați HKY din meniul derulant Subst Model. În mod ideal, ar trebui selectat un model de substituție care se potrivește cel mai bine datelor pentru fiecare partiție, dar aici, de dragul simplității, folosim HKY pentru toate partițiile. În continuare, selectați Empirical din meniul derulant Frequencies (Frecvențe). Acest lucru va fixa frecvențele la proporțiile observate în date (pentru fiecare partiție în parte, după ce vom decupla modelele de situs). Această abordare înseamnă că putem obține o ajustare bună la date fără a estima în mod explicit acești parametri. O facem aici doar pentru a face fișierele jurnal puțin mai scurte și mai ușor de citit în părțile ulterioare ale exercițiului.
Figura 7: Clonarea configurației de la un model de sit la altele.
În cele din urmă, bifați căsuța „estimate” pentru parametrul Substitution rate (Rata de substituție) și bifați căsuța Fix mean mutation rate (Fixarea ratei medii de mutație). Acest lucru va permite ca pentru fiecare partiție în parte să se estimeze ratele relative pentru modelele de situs fără legătură între ele (figura 6).
În cele din urmă, țineți apăsată tasta „shift” pentru a selecta toate modelele de situs din partea stângă și faceți clic pe OK pentru a clona setarea din non-codificare în 1stpos, 2ndpos și 3rdpos (figura 7). Treceți prin fiecare model de site, după cum puteți vedea, configurațiile lor sunt aceleași acum.
2.3 Setarea modelului de ceas
Postul următor este de a selecta fila Clock Models (Modele de ceas) în partea de sus a ferestrei principale. Acesta este locul unde selectăm modelul de ceas molecular. Pentru acest exercițiu, vom lăsa selecția la valoarea implicită a unui ceas molecular strict, deoarece aceste date sunt foarte asemănătoare cu un ceas și nu au nevoie de variația ratei între ramuri pentru a fi incluse în model.
Pentru a testa asemănarea cu ceasul, puteți (i) să executați analiza cu un model de ceas relaxat și să verificați cât de multă variație între rate este implicată de date (a se vedea coeficientul de variație pentru mai multe informații în acest sens), sau(ii) să efectuați o comparație de model între un ceas strict și unul relaxat folosind eșantionarea traseelor, sau (iii) să utilizați un model de ceas local aleatoriu care ia în considerare în mod explicit dacă fiecare ramură din arbore are nevoie de propria rată de ramificare.
2.4 Priors
Fila Priors permite specificarea priorilor pentru fiecare parametru din model. Selecțiile modelului făcute în filele „site model” și „clock model” au ca rezultat includerea diferiților parametri în model, iar aceștia sunt afișați în fila Priors (a se vedea figura 8).
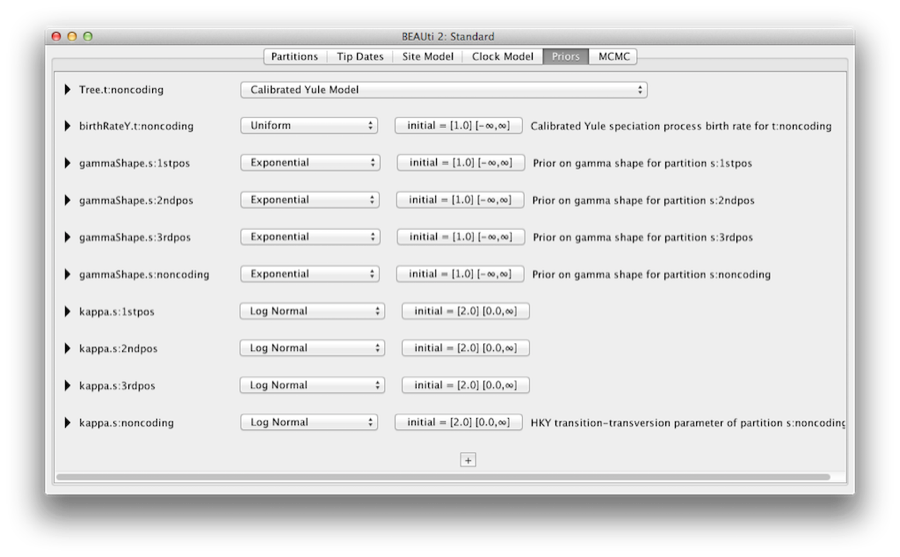
Figura 8: O captură de ecran a filei Priors din BEAUti.
Aici precizăm, de asemenea, că dorim să folosim modelul Yule calibrat ca prior al arborelui. Modelul Yule este un model simplu de speciație care este, în general, mai adecvat atunci când se iau în considerare secvențe de la specii diferite. Selectați Calibrated Yule Model (Modelul Yule calibrat) din meniul derulant Tree prior.
2.4.1 Definirea nodului de calibrare
Acum trebuie să precizăm o distribuție anterioară asupra nodului calibrat, pe baza cunoștințelor noastre despre fosile. Acest lucru este cunoscut sub numele de calibrarea arborelui nostru. Pentru a defini un prior suplimentar, apăsați butonul mic + de sub lista de priorități. Dacă acesta nu este vizibil în vizualizarea dvs., vă rugăm să derulați panoul până în partea de jos pentru a găsi butonul +. Veți vedea o fereastră de dialog care vă permite să definiți un subset de taxoni din arborele filogenetic. După ce ați creat un set de taxoni, veți putea adăuga ulterior informații de calibrare pentru cel mai recent commonancestor (MRCA) al acestuia.
Denumiți setul de taxoni prin completarea rubricii taxon set label (eticheta setului de taxoni). Numiți-l human-chimp, deoarece va conține taxonii pentru Homo sapiens și Pan. În lista de mai jos veți vedea taxonii disponibili. Selectați pe rând fiecare dintre cei doi taxoni și apăsați butonul săgeată > >. (Figura 9).Faceți clic pe OK și setul de taxoni nou-definit va fi adăugat în lista de stare anterioară.Deoarece acesta este un nod calibrat care urmează să fie utilizat împreună cu Stare anterioară Yule calibrată, trebuie să se aplice monofilia, așa că selectați caseta de selectare marcată Monophyletic. Acest lucru va constrânge topologia arborelui astfel încât gruparea om-călugăr să fie menținută monofilitică pe parcursul analizei MCMC.
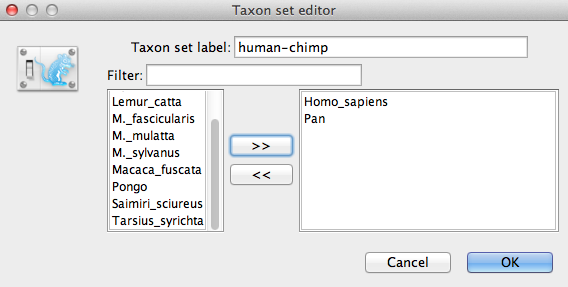
Figura 9: Editorul setului de taxoni în BEAUti.
Pentru a codifica informația de calibrare, trebuie să specificăm o distribuție pentru MRCA a omului-căpușă. selectați distribuția Log-normală din meniul derulant din dreapta umanului-căpușă nou adăugat.prior. Faceți clic pe triunghiul negru și va apărea un grafic al funcției de densitate de probabilitate, împreună cu parametrii pentru distribuția log-normală.Vom seta M = 1,78 și S = 0,085, ceea ce va specifica o distribuție centrată la aproximativ 6 milioane de ani cu o deviație standard de aproximativ 0,5 milioane de ani. Acest lucru va da un interval central de probabilitate de 95% care acoperă 5-7 Mya. Aceasta corespunde aproximativ cu estimarea consensuală actuală a datei celui mai recent strămoș comun al oamenilor și cimpanzeilor (figura 10).
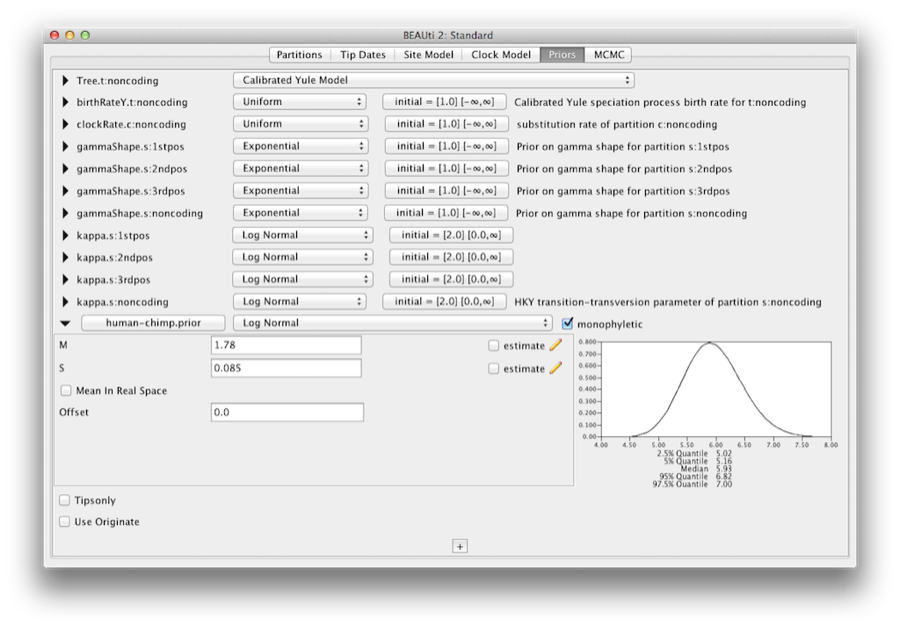
Figura 10: O captură de ecran a opțiunilor de calibrare a priorității din panoul Priors din BEAUti.
Ar trebui să ne convingem că prioritățile afișate în panoul priors reflectă cu adevărat informațiile anterioare pe care le avem despre parametrii modelului. În cele din urmă, vom specifica, de asemenea, unele priori difuze „neinformative”, dar adecvate, asupra ratei globale a ceasului molecular (clockRate) și a ratei de speciație (birthRateY) din cadrul priorității arborelui Yule. Pentru fiecare dintre ele, selectați Gamma din meniul derulantși, cu ajutorul butonului săgeată, extindeți vizualizarea pentru a dezvălui parametrii prioritățiiGamma. Atât pentru rata ceasului, cât și pentru rata de natalitate Yule, setați parametrul Alpha(shape) la 0,001 și parametrul Beta (scale) la 1000 (figura 11).
În mod implicit, fiecare dintre parametrii de formă gamma are o priordistribuție exponențială cu o medie de 1. Acest lucru implică (a se vedea figura 3.7) că ne așteptăm la o variație oarecare. În mod implicit, parametrii kappa pentru modelul HKY au o distribuție anterioară lognormală(1,1,25), care este în general în concordanță cu dovezile empirice cu privire la intervalul de valori realiste ale deviațiilor de tranziție/transversie. Aceste priorități implicite sunt păstrate deoarece sunt adecvate pentru această analiză particulară.
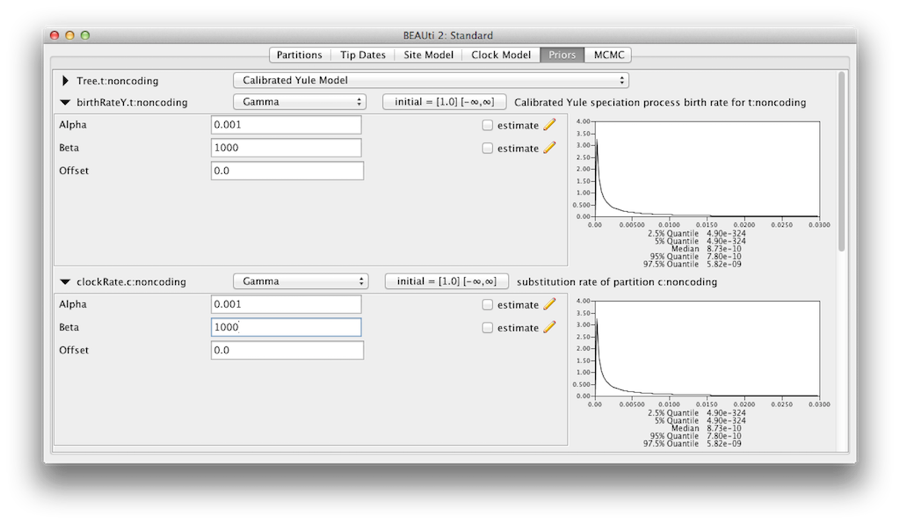
Figura 11: Gamma prior.
2.5 Setarea opțiunilor MCMC
Postul următor, MCMC, oferă mai multe setări generale pentru a controla durata execuției MCMC și numele fișierelor.
În primul rând avem Chain Length (Lungimea lanțului). Acesta este numărul de pași pe care MCMC îi va face în lanț înainte de a se termina. Cât de lungă ar trebui să fie aceasta depinde de dimensiunea setului de date, de complexitatea modelului și de calitatea răspunsului necesar. Valoarea implicită de 10 000 000 000 este complet arbitrară și trebuie ajustată în funcție de dimensiunea setului de date. Pentru acest set de date, să stabilim lungimea lanțului la 6.000.000.000, deoarece acest lucru se va executa rezonabil de repede pe majoritatea calculatoarelor moderne (câteva minute).
Câmpului Store Every determină cât de des este stocată starea în fișier. Stocarea periodică a stării este utilă pentru situațiile în care mediul de calcul nu este foarte fiabil și o execuție BEAST poate fi întreruptă. Faptul de a avea o copie stocată a stării recente vă permite să reluați lanțul în loc să reporniți de la început, astfel încât nu trebuie să treceți din nou prin burn-in. câmpul Pre Burnin specifică numărul de eșantioane care nu sunt înregistrate chiar la începutul analizei. Noi lăsăm câmpurile Store Every și Pre Burnin setate la valorile lor implicite. Mai jos sunt prezentate detaliile fișierelor jurnal. Fiecare dintre ele poate fi extins făcând clic pe triunghiul negru.
Opțiunile următoare specifică frecvența cu care valorile parametrilor din lanțul Markov ar trebui să fie afișate pe ecran și înregistrate în fișierul jurnal.Ieșirea pe ecran servește pur și simplu la monitorizarea progresului programului și poate fi setată la orice valoare (deși, dacă este setată la o valoare prea mică, cantitatea mare de informații afișate pe ecran va încetini de fapt programul). Pentru fișierul jurnal, valoarea trebuie să fie stabilită în funcție de lungimea totală a lanțului. O eșantionare prea frecventă va avea ca rezultat fișiere foarte mari, cu puține beneficii suplimentare în ceea ce privește acuratețea analizei. Dacă eșantionarea este prea puțin frecventă, fișierul jurnal nu va înregistra suficiente informații despre distribuția parametrilor. Probabil că doriți să încercați să nu stocați mai mult de 10.000 de eșantioane, astfel încât acest lucru ar trebui să fie egal cu nu mai puțin de lungimea lanțului / 10.000.
Pentru acest exercițiu vom seta frecvența jurnalului de urmărire și a jurnalului de arbori la 1.000 și a jurnalului de ecran la 10.000. De asemenea, specificați Primates.log ca nume de fișier pentru fișierul jurnal de urmărire și Primates.trees ca nume de fișier pentru fișierul jurnal de arbori.Asigurați-vă că fișierul cu numele de fișier al jurnalului de ecran este lăsat gol, altfel jurnalul de ecran nu va fi scris pe ecran.
- Dacă utilizați sistemul de operare Windows, vă sugerăm să adăugați sufixul .txt la ambele fișiere (deci, Primates.log.txt și Primates.trees.txt), astfel încât Windows să le recunoască drept fișiere text.
2.6 Generarea fișierului BEAST XML
Suntem acum gata să creăm fișierul BEAST XML. Pentru a face acest lucru,selectați opțiunea Save (Salvare) din meniul File (Fișier). Bifați prioritățile implicite și salvați fișierul cu un nume adecvat (de obicei, terminăm numele de fișier cu .xml, de exemplu, Primates.xml).Acum suntem gata să rulăm fișierul prin BEAST.
3 Rularea BEAST
Figura 12: O captură de ecran a BEAST.
Acum rulați BEAST și când vă cere un fișier de intrare, furnizați fișierul XML nou creat ca intrare. BEAST va rula apoi până când se terminăreportarea informațiilor pe ecran. Fișierele cu rezultatele efective sunt salvate pe disc în aceeași locație ca și fișierul de intrare. Ieșirea pe ecran va arăta cam așa:
BEAST v2.2.0, 2002-2014 Bayesian Evolutionary Analysis Sampling Trees Designed and developed byRemco Bouckaert, Alexei J. Drummond, Andrew Rambaut and Marc A. Suchard Department of Computer Science University of Auckland [email protected] [email protected] Institute of Evolutionary Biology University of Edinburgh [email protected] David Geffen School of Medicine University of California, Los Angeles [email protected] Downloads, Help & Resources: http://beast2.org/ Source code distributed under the GNU Lesser General Public License: http://github.com/CompEvol/beast2 BEAST developers: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Denise Kuehnert, Philippe Lemey, Wai Lok Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Chieh-Hsi Wu, Walter Xie Thanks to: Roald Forsberg, Beth Shapiro and Korbinian StrimmerRandom number seed: 77712 taxa898 sites413 patternsTreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4TreeLikelihood uses beast.evolution.likelihood.BeerLikelihoodCore4===============================================================================Citations for this model:Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10(4): e1003537Heled J, Drummond AJ (2012) Calibrated Tree Priors for Relaxed Phylogenetics and Divergence Time Estimation. Systematic Biology 61(1):138-149.Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.===============================================================================Writing file /Primates.logWriting file /Primates.trees Sample posterior ESS(posterior) likelihood prior 0 -7924.3599 N -7688.4922 -235.8676 -- 10000 -5529.0700 2.0 -5459.1993 -69.8706 -- 20000 -5516.8159 3.0 -5442.3372 -74.4786 -- 30000 -5516.4959 4.0 -5439.0839 -77.4119 -- 40000 -5521.1160 5.0 -5445.6047 -75.5113 -- 50000 -5520.7350 6.0 -5444.6198 -76.1151 -- 60000 -5512.9427 7.0 -5439.2561 -73.6866 2m39s/Msamples 70000 -5513.8357 8.0 -5437.9432 -75.8924 2m39s/Msamples ... 5990000 -5516.6832 474.6 -5442.5945 -74.0886 2m40s/Msamples 6000000 -5512.3802 472.2 -5440.8928 -71.4874 2m40s/MsamplesOperator Tuning #accept #reject total prob.accScaleOperator(treeScaler.t:tree) 0.703 39935 174155 214090 0.187 ScaleOperator(treeRootScaler.t:tree) 0.644 37329 177166 214495 0.174 Uniform(UniformOperator.t:tree) 479419 1668915 2148334 0.223 SubtreeSlide(SubtreeSlide.t:tree) 9.922 272787 801404 1074191 0.254 Exchange(narrow.t:tree) 744 1074261 1075005 0.001 Exchange(wide.t:tree) 9 214594 214603 0.000 WilsonBalding(WilsonBalding.t:tree) 4 214548 214552 0.000 ScaleOperator(KappaScaler.s:noncoding) 0.352 1739 5375 7114 0.244 DeltaExchangeOperator(FixMeanMutationRatesOperator) 0.425 17277 126203 143480 0.120 ScaleOperator(gammaShapeScaler.s:noncoding) 0.375 1729 5428 7157 0.242 ScaleOperator(CalibratedYuleBirthRateScaler.t:tree) 0.245 58005 156128 214133 0.271 ScaleOperator(StrictClockRateScaler.c:clock) 0.706 50080 164952 215032 0.233 UpDownOperator(strictClockUpDownOperator.c:clock) 0.589 50809 163882 214691 0.237 ScaleOperator(KappaScaler.s:1stpos) 0.44 1816 5388 7204 0.252 ScaleOperator(gammaShapeScaler.s:1stpos) 0.42 1927 5129 7056 0.273 ScaleOperator(KappaScaler.s:2ndpos) 0.332 1964 5301 7265 0.270 ScaleOperator(gammaShapeScaler.s:2ndpos) 0.303 2033 5177 7210 0.282 ScaleOperator(KappaScaler.s:3rdpos) 0.505 1424 5860 7284 0.195 ScaleOperator(gammaShapeScaler.s:3rdpos) 0.267 1569 5536 7105 0.221 Total calculation time: 964.067 seconds
Rețineți că la început există unele informații utile cu privire la alinieri și la probabilitățile arborelui care sunt folosite. De asemenea, toate citatele relevante pentru analiză sunt menționate la începutul rulării, care pot fi ușor copiate în manuscrisele care raportează despre analiză. Urmează apoi raportarea lanțului,care oferă un feedback în timp real cu privire la progresul lanțului.
La sfârșit, se tipărește o analiză a operatorilor, care enumeră toți operatorii utilizați în analiză împreună cu cât de des a fost încercat, acceptat și respins operatorul (a se vedea coloanele #total, #accept și, respectiv, #reject). Rata de acceptare este proporția de cazuri în care un operator este acceptat atunci când este selectat pentru a face o propunere. În general, o rată de acceptare ridicată, de exemplu peste 0,5, indică faptul că propunerile sunt conservatoare și nu explorează eficient spațiul parametrilor. Pe de altă parte, o rată de acceptare scăzută indică faptul că propunerile sunt prea agresive și rezultă aproape întotdeauna într-o stare care este respinsă din cauza posteriorului său scăzut.Atât ratele de acceptare prea mari, cât și cele prea mici au ca rezultat valori ESS scăzute. O rată de acceptare de 0,234 este ținta (pe baza dovezilor foarte limitate furnizate de ) pentru mulți (dar nu toți) operatorii implementați în BEAST.
Câțiva operatori au un parametru de reglare, de exemplu factorul de scară al parametrului ascale. În cazul în care rata finală de acceptare nu este aproape de țintă, BEASTsugerează o nouă valoare pentru parametrul de reglare, care este imprimată în analiza operatorului. În acest caz, toate ratele de acceptare sunt bune pentru operatorii care au parametri de reglare. Operatorii fără parametri de reglare includ operatorii wideexchange și Wilson-Balding pentru această analiză. Amândoi acești operatori încearcă să schimbe topologia arborelui cu pași mari, dar deoarece datele susțin în mod covârșitor o singură topologie, aceste propuneri radicale sunt aproape întotdeauna respinse.
4 Analiza rezultatelor
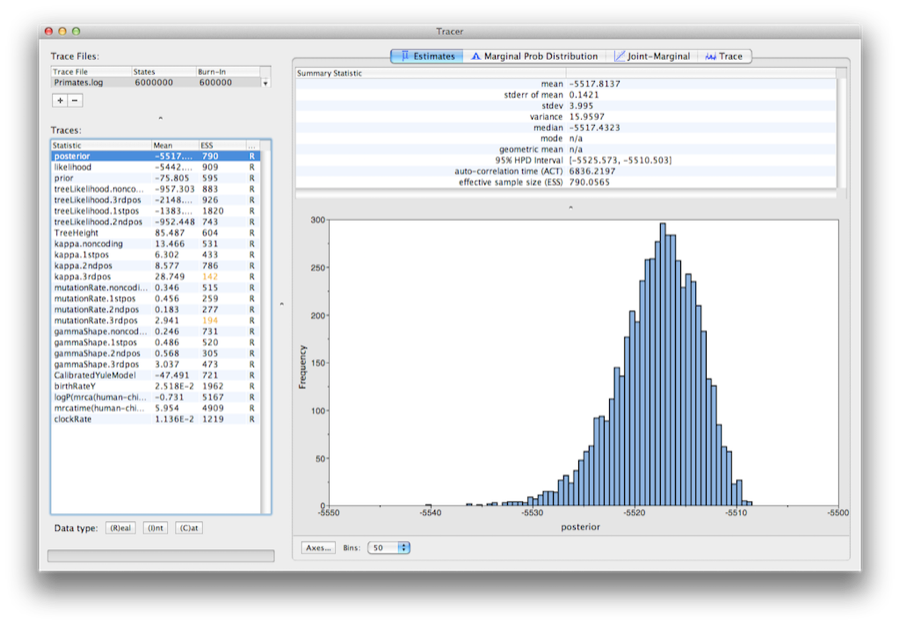
Figura 13: O captură de ecran a programului Tracer v1.6.
Executați programul numit Tracer pentru a analiza rezultatele obținute de BEAST. Când fereastra principală s-a deschis, alegeți Import Trace File… din meniul File și selectați fișierul pe care BEAST l-a creat numit Primates.log (Figura 13).
Rețineți că MCMC este un algoritm stocastic, astfel încât numerele reale nu vor fi exact aceleași cu cele descrise în figură.
În partea stângă se află o listă a diferitelor cantități pe care BEAST le-a înregistrat în fișier. Există urme pentru posterior (acestaeste logaritmul natural al produsului dintre probabilitatea arborelui și densitatea anterioară) și parametrii continui. Selectarea unei urme din stânga aduce analize pentru această urmă în partea dreaptă, în funcție de fila care este selectată. La prima deschidere, este selectată urma „posterioară” și diverse statistici ale acestei urme sunt afișate în fila Estimates (Estimări). în partea dreaptă sus a ferestrei se află un tabel cu statisticile calculate pentru urma selectată.
Selectați parametrul clockRate din lista din stânga pentru a observa rata medie de evoluție (calculată ca medie pe întregul arbore și pe toate siturile). Tracer va trasa o histogramă (marginală posterioară) pentru statistica selectată și vă va oferi, de asemenea, statistici sumare, cum ar fi media și mediana. HPD 95% reprezintă intervalul cu cea mai mare densitate posterioară și reprezintă cel mai compact interval pentru parametrul selectat care conține 95% din probabilitatea posterioară. Acesta poate fi considerat, în linii mari, ca un analog bayesian al unui interval de încredere. Parametrul TreeHeight oferă distribuția marginală posterioară a vârstei rădăcinii întregului arbore.
Selectați parametrul TreeHeight și apoi faceți Ctrl-clic pe mrcatime(human-chimp) (Command-clic pe Mac OS X). Acest lucru va afișa o vizualizare a vârstei rădăcinii și a MRCA de calibrare pe care am specificat-o mai devreme în BEAUti. Puteți verifica faptul că divergența pe care am folosit-o pentru a calibra arborele(mrcatime(human-chimp)) are o distribuție posterioară care se potrivește cu distribuția anterioară pe care am specificat-o (Figura 14).
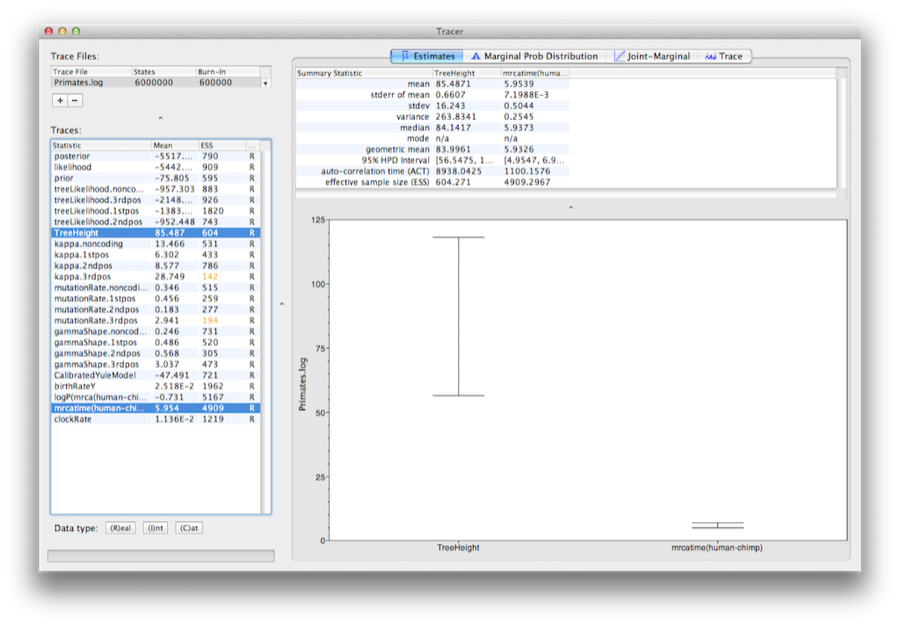
Figura 14: O captură de ecran a intervalelor HPD de 95% ale înălțimii rădăcinii și ale MRCA-ului specificat de utilizator (human-chimp) în Tracer.
5 Estimări marginale posterioare
Pentru a afișa ratele relative pentru cele patru partiții, selectați parametrul mutationRate pentru fiecare dintre cele patru partiții și selectați fila marginal density (densitate marginală) în Tracer.Figura 15 prezintă densitățile marginale pentru ratele relative de substituție. Graficul arată că pozițiile de codon 1 și 2 au rate substanțial diferite (0,456versus 0,183) și ambele sunt mult mai lente decât poziția de codon 3, cu o rată relativă de2,941. Partiția necodificatoare are o rată intermediară între pozițiile de codon 1 și 2 (0,346). Împreună, acest rezultat sugerează o selecție purificatoare puternică atât în regiunile codificatoare, cât și în cele necodificatoare ale alinierii.
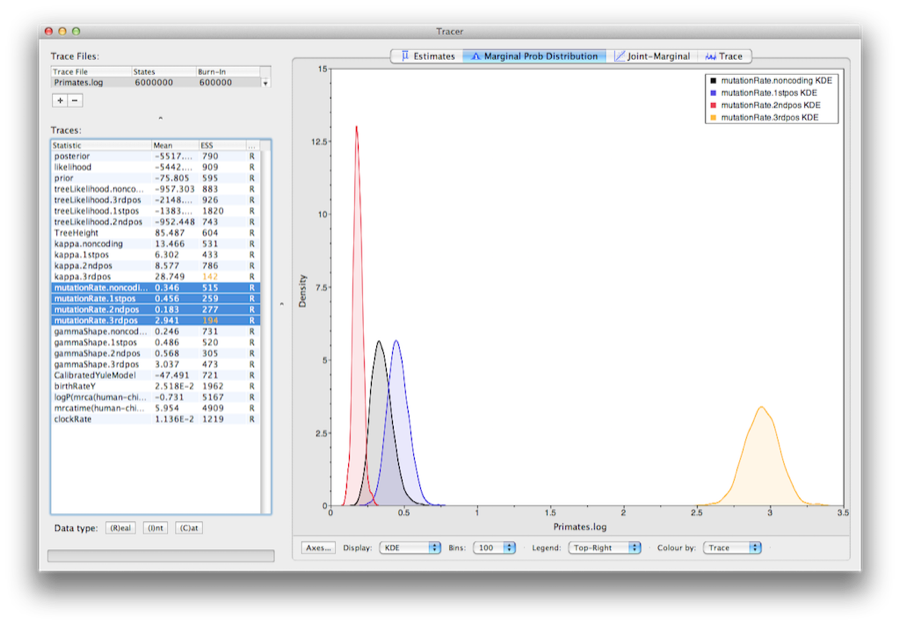
Figura 15: O captură de ecran a densităților posterioare marginale ale ratelor relative de substituție ale celor patru partiții (în raport cu rata medie ponderată pe site).
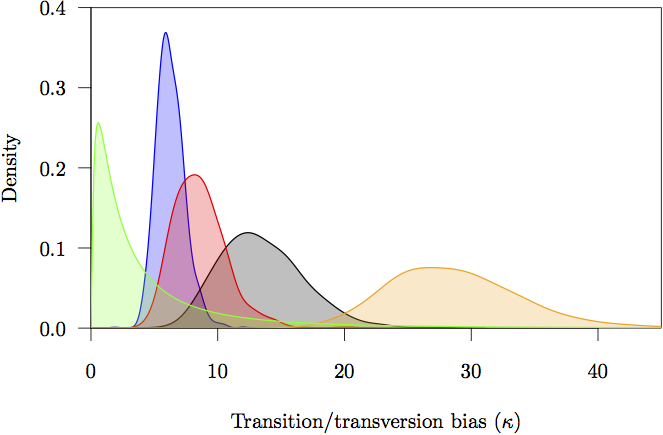
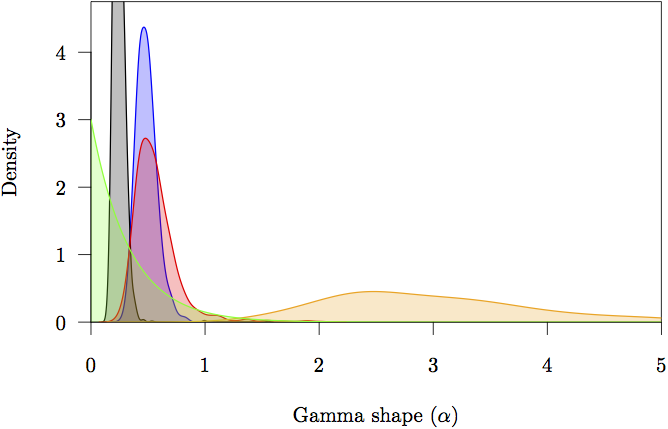
Figura 16: Densitățile marginale anterioare și posterioare pentru parametrii de formă (α). Prioritatea este în gri. Este, de asemenea, prezentată estimarea densității posterioare pentru fiecare partiție: pozițiile necodificatoare (portocaliu) și prima (roșu), a doua (verde) și a treia (albastru) poziție a codonului.
Figura 17: Densitățile marginale anterioare și posterioare pentru parametrii de polarizare a tranziției/transversiei (κ). Prioritatea este în gri. Este, de asemenea, prezentată estimarea densității posterioare pentru fiecare partiție: pozițiile necodificatoare (portocaliu) și prima (roșu), a doua (verde) și a treia (albastru) poziție a codonului.
Întrebări
Care este rata estimată a evoluției moleculare pentru acest arbore genic (includeți intervalul HPD de 95%)?
Ce surse de eroare include această estimare?
Câți ani are rădăcina arborelui (dați media și intervalul HPD de 95%)?
6 Obținerea unei estimări a arborelui filogenetic
BEAST produce, de asemenea, un eșantion posterior de arbori cronologici filogenetici împreună cu eșantionul său de estimări ale parametrilor. Acestea trebuie să fie rezumate cu ajutorul programului TreeAnnotator. Acesta va lua setul de arbori și îl va găsi pe cel mai bine susținut. Acesta va adnota apoi acest arbore rezumativ reprezentativ cu vârstele medii ale tuturor nodurilor și intervalele HPD de 95% corespunzătoare. Se va calcula, de asemenea, probabilitatea posterioară a cladelor pentru fiecare nod. Rulați programul TreeAnnotator și configurați-l așa cum este ilustrat în figura 18.
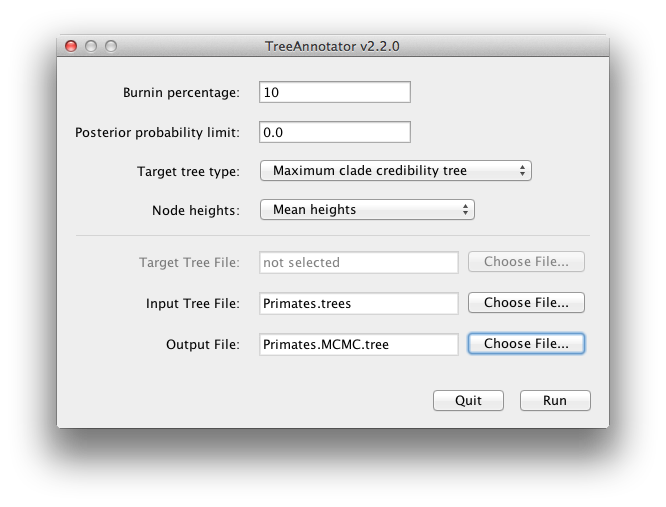
Figura 18: O captură de ecran a programului TreeAnnotator.
Arboretul este numărul de arbori care trebuie eliminați de la începutul eșantionului. Spre deosebire de Tracer, care specifică numărul de pași ca burnin, în TreeAnnotator trebuie să specificați numărul real de arbori. Pentru această execuție, ați specificat o lungime a lanțului de 6 000 000 de pași de eșantionare la fiecare 1 000 de pași. Astfel, fișierul trees va conține 6.000 de arbori și, prin urmare, pentru a specifica un burnin de 10% în câmpul de text superior.
Opțiunea Posterior probability limit specifică o limită astfel încât, dacă un nod se găsește la o frecvență mai mică decât această frecvență în eșantionul de arbori (adică are o probabilitate posterioară mai mică decât această limită), acesta nu va fi adnotat. Valoarea implicită de 0,5 înseamnă că numai nodurile observate în majoritatea arborilor vor fi notate. Setați această valoare la zero pentru a adnota toate nodurile.
The Target tree type specifică topologia arborelui care va fi adnotată. Puteți alege un arbore specific dintr-un fișier sau puteți cere TreeAnnotator să găsească un arbore în eșantionul dumneavoastră. opțiunea implicită, Maximum clade credibility tree, găsește arborele cu cel mai mare produs al probabilității posterioare a tuturor nodurilor sale.
Pentru înălțimea nodurilor, opțiunea implicită este Common Ancestor Heights, care calculează înălțimea unui nod ca medie a timpului MRCA al tuturor perechilor de noduri din cladă. În cazul arborilor cu o mare incertitudine în topologie și, prin urmare, cu multe clade cu suport scăzut, unele alte metode pot avea ca rezultat arbori cu lungimi de ramură negative. În această analiză, suportul pentru toate cladele din arborele rezumativ este foarte mare, astfel încât acest lucru nu reprezintă o problemă în acest caz.Alegeți înălțimi medii pentru înălțimile nodurilor. Acest lucru stabilește înălțimile (vârstele) fiecărui nod din arbore la înălțimea medie din întregul eșantion de arbori pentru acea cladă.
Pentru fișierul de intrare, selectați fișierul de arbori pe care BEAST l-a creat și selectați un fișier de ieșire (aici l-am numit Primates.MCC.tree). Acum apăsați Run și așteptați ca programul să se termine.
7 Vizualizarea estimării arborelui
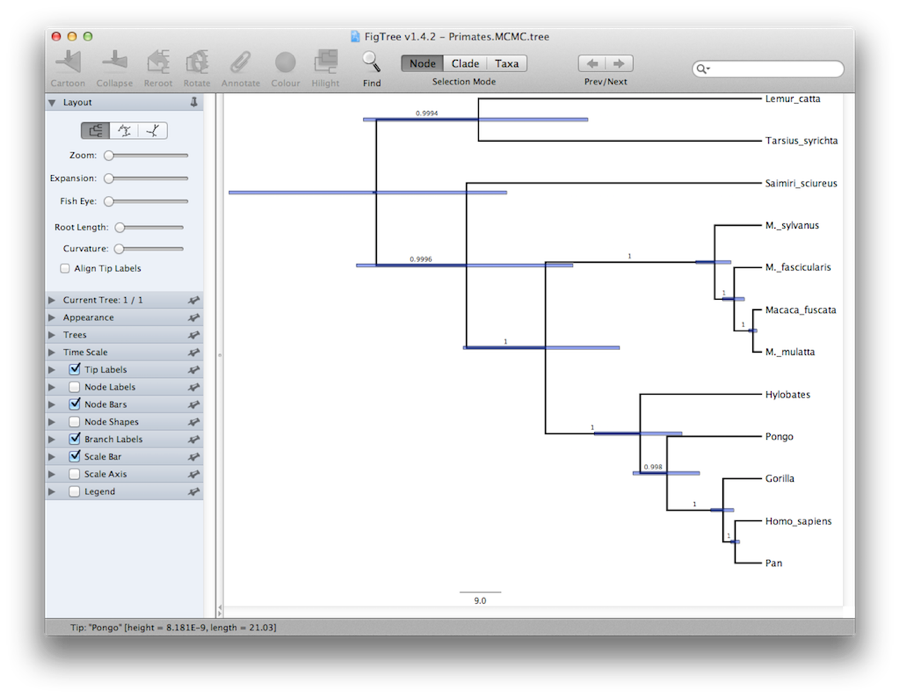
În cele din urmă, putem vizualiza arborele într-un alt program numit FigTree. Rulați acest program și deschideți fișierul Primates.MCC.tree cu ajutorul comenzii Open din meniul File. Arborul ar trebui să apară. puteți încerca acum să selectați unele dintre opțiunile din panoul de control din stânga. În primul rând, cheltuiți opțiunea Trees din panou și bifați Order nodes și alegeți Ordering by decreasing. Încercați să selectați Node Bars pentru a obține bare de eroare privind vârsta nodurilor. De asemenea, activați Branch Labels (Etichete de ramură) și selectați posterior pentru ca acesta să afișeze probabilitatea posterioară pentru fiecare nod. Dacă folosiți un model de ceas care nu este strict, atunci la rubrica Appearance (Aspect) puteți, de asemenea, să îi spuneți lui FigTree să coloreze ramurile în funcție de rată. ar trebui să obțineți ceva similar cu figura 19.
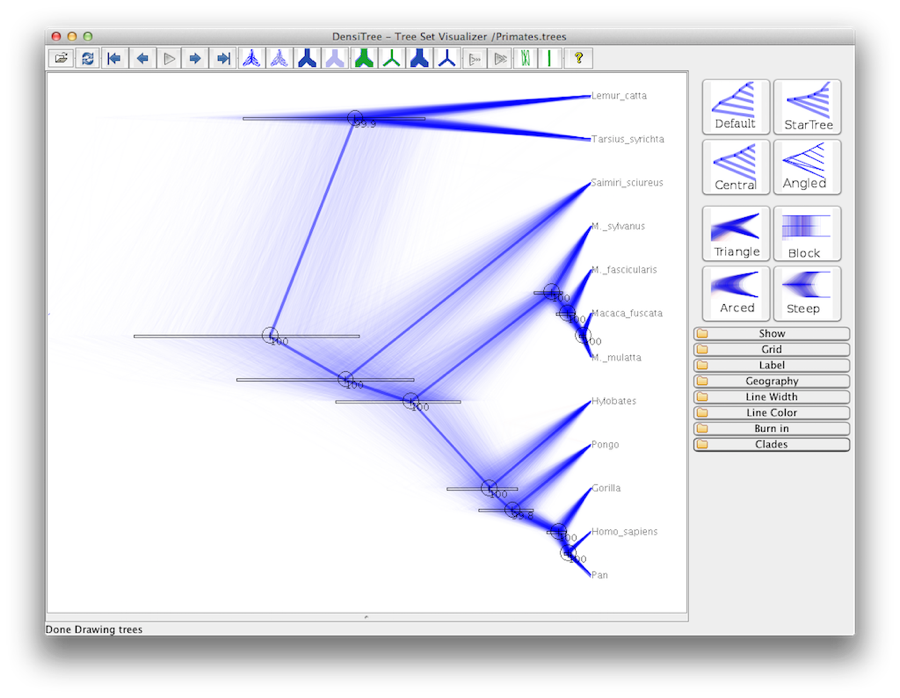
Figura 19: O captură de ecran a FigTree și DensiTree.
O vizualizare alternativă a arborelui poate fi realizată cu DensiTree, care face parte din Fiara 2. Avantajul lui DensiTree este că este capabil să vizualizeze atât incertitudinea în înălțimile nodurilor, cât și incertitudinea în topologie. pentru acest set de date particular, topologia dominantă este prezentă în peste 99% din eșantioane. Așadar, concluzionăm că această analiză are ca rezultat un consens foarte ridicat în ceea ce privește topologia (Figura 19).
Întrebări
- Ratele de evoluție diferă substanțial între diferitele linii genetice din arbore?
- DensiTree are o bară de clade (Menu Window/View clade toolbar) pentru a afișa informații despre clade.
Care este suportul pentru clade?
- Puteți naviga prin topologiile din DensiTree folosind meniul Browse.Cea mai populară topologie are un suport de peste 99%.
Care este suportul pentru a doua cea mai populară topologie?
- În meniul de ajutor, DensiTree arată câteva informații.
Câte topologii sunt în setul de arbori?
8 Compararea rezultatelor dvs. cu cele anterioare
Este o idee bună să rulați din nou analiza în timp ce eșantionați din a priori pentru a vă asigura că interacțiunile dintre a priori nu vă afectează informațiile anterioare. Interacțiunea dintre priori poate fi problematică mai ales atunci când se folosesc calibrări, deoarece înseamnă să se pună mai multe priori pe arbore.
Utilizând BEAUti, configurați aceeași analiză, dar în cadrul opțiunilor MCMC, selectați opțiunea Sample from prior only. Acest lucru vă va permite să vizualizați întreaga distribuție prioritară în absența datelor de secvență. Rezumați arborii din distribuția priordistribuția completă și comparați rezumatul cu arborele rezumativ posterior.
Stimarea timpului de divergență folosind „datarea nodurilor” de tipul descris în acestcapitol a fost aplicată pentru a răspunde la o varietate de întrebări diferite în ecologie și evoluție. De exemplu, datarea nodurilor cu ajutorul fosilelor a fost utilizată pentru a determina diversitatea speciilor de cicade , pentru a analiza rata de evoluție la plantele cu flori și pentru a investiga originea cianobacteriilor din deșerturile calde și reci .
Justin Bahl, Maggie CY Lau, Gavin JD Smith, Dhanasekaran Vijaykrishna, S CraigCary, Donnabella C Lacap, Charles K Lee, R Thane Papke, Kimberley AWarren-Rhodes, Fiona KY Wong, et al, Ancient origins determine globalbiogeography of hot and cold desert cyanobacteria, Nature communications2 (2011), 163. Alexei J Drummond și Marc A Suchard, Bayesian random local clocks, orone rate to rule them all, BMC biology 8 (2010), nr. 1, 114. A Gelman, G Roberts și W Gilks, Efficient metropolis jumping hules,Bayesian statistics 5 (1996), 599-608. Joseph Heled și Alexei J Drummond, Calibrated tree priors for relaxedphylogenetics and diverence time estimation, Syst Biol 61 (2012),nr. 1, 138-49. NS Nagalingum, CR Marshall, TB Quental, HS Rai, DP Little și S Mathews, Recent synchronous radiation of a living fossil, Science 334(2011), nr. 6057, 796-799. Michael S Rosenberg, Sankar Subramanian și Sudhir Kumar, Patterns oftransitional mutation biases within and among mammalian genomes, Molecularbiology and evolution 20 (2003), nr. 6, 988-993. Stephen A Smith și Michael J Donoghue, Rates of molecular evolution arelinked to life history in flowering plants, science 322 (2008), nr. 5898, 86-89.
Acest document a fost tradus din LATEX de cătreHEVEA.